势能面扫描

乙烷及乙烷取代物绕C-C键旋转势能面扫描
方法步骤:(内坐标法scan):
1.同上用gaussianview做输入文件:分别构建乙烷,乙醇,乙二醇分子,保存为11.gjf 1
2.gjf 1
3.gjfG03输入文件格式。
2.修改gaussianview生成的内坐标:分别用gaussianview打开11.gjf 12.gjf 1
3.gjf并显示原子编号,根据图像对应坐标修改,使其只有一个二面角的改变可引起乙烷两个甲基相对旋转。如下所示乙烷;
0 1
C
H 1 B1
H 1 B2 2 A1
H 1 B3 3 A2 2 D1
C 1 B4 4 A3 3 D2
H 5 B5 1 A4 4 D3
H 5 B6 1 A5 4(6)D4
H 5 B7 1 A6 4(7 )D5
B1 1.07000000
B2 1.07000000
B3 1.07000000
B4 1.54000000
B5 1.07000000
B6 1.07000000
B7 1.07000000
A1 109.47120255
A2 109.47121829
A3 109.47121829
A4 109.47120255
A5 109.47120255
A6 109.47123134
D1 -120.00000060
D2 120.00003407
D3 179.98890060
D4 59.88891539 (115.)
D5 179.88890799 (115.)
然后进行正常优化和频率分析(#hf/3-21g opt freq )。
3.建立扫描文件:同前判断11.OUT .12.OUT 13.OUT优化结果,2个normal4个yes,无虚频。然后分别在11.OUT .12.OUT 13.OUT中找到Final structure in terms of initial Z-matrix:把对应坐标粘贴建立扫描文件11scan.gjf.12scan.gjf.13scan.gjf。
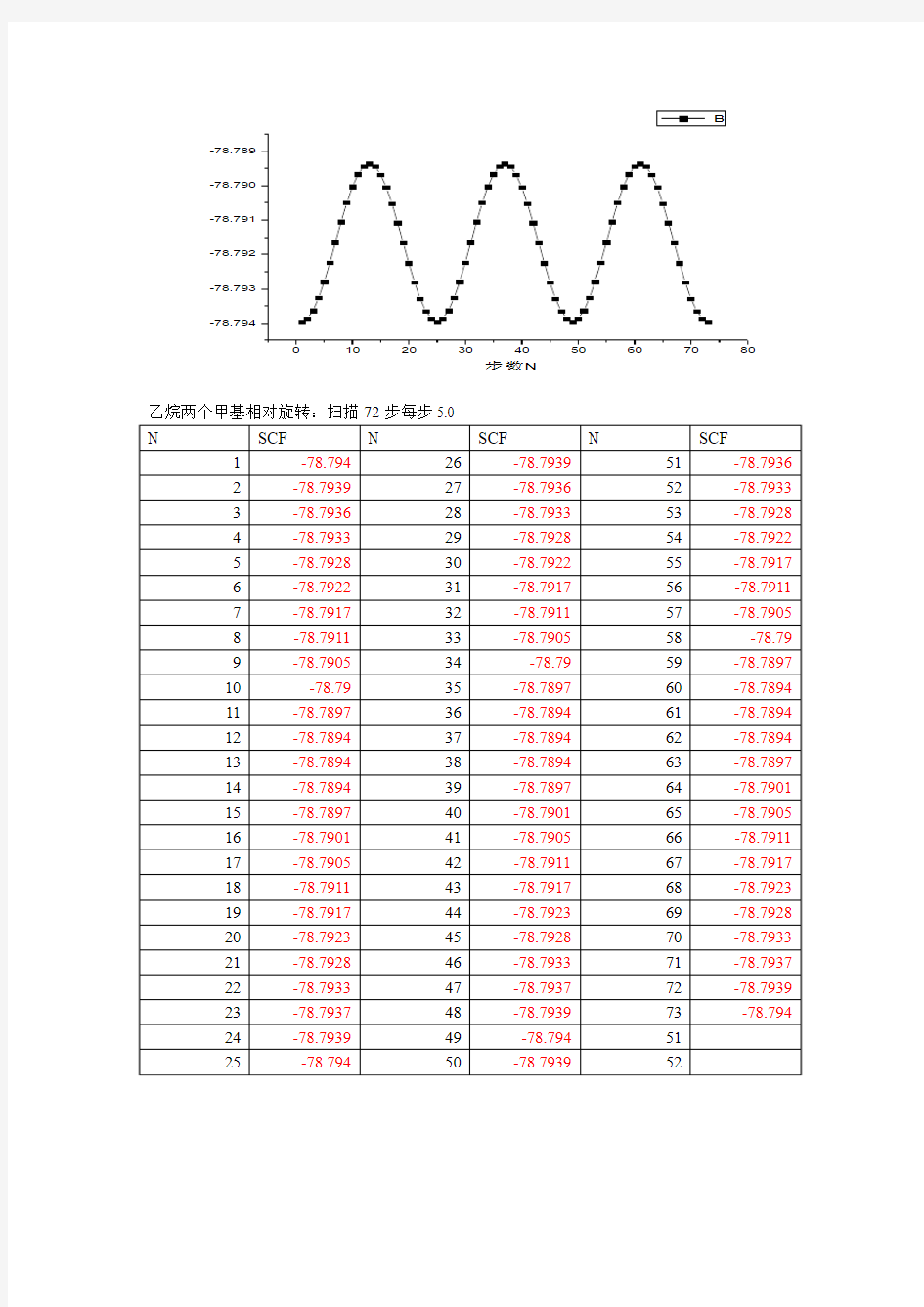
4. 扫描结果;用Summary of the potential surface scan处所对应的SCF值和步数origin6.0作图。
能量(A .U )
步数N
乙烷两个甲基相对旋转:扫描72步每步5.0
能量(A .U )
步数N
能量(A .U )
步数N
乙二醇分子如上述方式旋转: 扫描72步每步5.0
第6章塑性应力-应变关系01分析
第6章 塑性应力-应变关系 在20世纪50年代,经典塑性理论有了很大的发展,表现在:(1)极限分析的基本定理(Drucker 等,1952);(2)Drucker 假设或稳定材料的定义(Drucker ,1951);(3)正交性条件的概念或关联流动法则(Drucker ,1960)等的建立和发展。理想塑性体的极限分析理论产生了能更直接地估计结构和土体承载力的实际方法(Chen ,1982,Chen 和Liu ,1990)。稳定材料的概念提供了一个统一的方法和塑性体的应力-应变关系的广义观点。正交性条件的概念提供了塑性应力-应变关系的屈服准则或加载函数之间的必要联系。所有的这些进展导出了金属塑性经典理论严格的基础,也为后来土体、岩石和混凝土类的其他材料的更复杂的塑性理论发展打下了基础(Chen 和Han ,1988,Chen 和Mizuno ,1990)。 6.1 加载准则 在应力空间上的屈服面确定了当前的弹性区的边界。如果一个应力点在屈服面的里面,就称之为弹性状态而且只有弹性特性;如果一个应力点在屈服面上,其应力状态为塑性状态,产生弹性或者弹塑性特性。 在数学上,弹性状态和塑性状态作如下定义: 0 势能面相交规则 ————————————————————————————————作者:————————————————————————————————日期: 1.7 势能面相交规则 一个分子体系可以有不同的电子态,非简并态具有不同的能级,因而有不同的势能面,这些势能面都是(3N-6)/(3N-5)个独立坐标的函数. 不同势能面有可能相交,当两个势能面发生交叉时,两能级将出现简并,习惯上称之为“偶然简并”. 在势能面相交区域,分子常常会表现出一些特殊的动态学行为,因此有必要研究势能面的相交规则. 先讨论双原子分子的势能曲线,此时,我们有如下的相交规则: 对称性(包括自旋和空间)相同的态的势能曲线不能相交,对称性不同的态 的势能曲线可以相交. 简要解释一下上述规则. 我们知道,势能曲线是电子哈密顿量的本征能量曲 线,相应的本征函数应具有两种对称性,一是自旋对称性,即电子波函数应该是自旋算符的本征函数,总自旋算符有确定值S ,通常用(21)S +表示电子波函数的自旋多重度. 此外,电子波函数还应具有分子所属点群的对称性,即它应当是分子所属点群不可约表示的基. 上述规则意味着,当两个电子态波函数具有相同的自旋多重度并属于分子点群同一不可约表示时,相应的势能曲线不能相交,称之为避免交叉. 现在来证明这一规则. 双原子分子具有h D ∞(同核)或h C ∞(异核)对称性. 设1(21) () 1(,)S R q +λv Ψ和2(21)()2(,)S μR q +v ψ为某一双原子分子的两个正交归一化 的非简并电子态波函数,分别为点群的λ和μ不可约表示的基,自旋多重度分别为1(21)S +和2(21)S +,相应的势能曲线分别记作)(1R E 和)(2R E . 如图(1.14) 所示。 图1.14 势能曲线相交 ADF教程:如何进行势能面扫描 两个例子:(1)乙醇中羟基的拉伸;(2)乙醇中羟基的转动。 乙醇中羟基的拉伸 参数设置 Preset选择任务类型为Linear Transit,并选择合适的基组和泛函: 选中要扫描的键长,本例中为拉伸羟基与乙基的距离,因此如下图所示,按住shift键,选择C和O,然后在Model > Geometry Constraints and Scan中点击对应的+,表示要扫描C和O原子之间的距离: 之后设置具体的扫描范围。其中起始值为当前值。并设置扫描多少个点,本例中为10个点。并且扫描的参数也可以不止一个,可以多个同步扫描。 保存任务并计算。 结果查看 ADF LOGO > Movie > Graph可以看到整个拉伸过程,能量的变化: 注意,我们虽然扫描10个点,但每个点,结构如何?程序需要进行能量最小化才知道,也就是限制键长为设定的10个渐变数值,然后优化其它原子的位置,使得能量达到最低、最稳定的结构。 View > Converged Geometries Only,这样只显示优化之后的结构,也就是实际拉伸过程,得到的10个结构,当然对应的能量也在右边显示出来了: 之后选中C和O,Graph>Distance,Angle,Dihedral,之后点击Graph>Curve on X Axes,这样就显示能量随键长的变化: Graph>Save as XY可以保存能量与键长的函数曲线xy坐标值。 乙醇中羟基的转动 参数设置与上面类似,不过这里我们希望转动-OH,因此实际上是扫描H-O-C-C的二面角。因为在ADFinput中,依次选中这4个原子,设置二面角的变化范围: 其它与上面的操作完全一样。本教程到此结束。 1.7 势能面相交规则 一个分子体系可以有不同的电子态,非简并态具有不同的能级,因而有不同的势能面,这些势能面都是(3N-6)/(3N-5)个独立坐标的函数. 不同势能面有可能相交,当两个势能面发生交叉时,两能级将出现简并,习惯上称之为“偶然简并”. 在势能面相交区域,分子常常会表现出一些特殊的动态学行为,因此有必要研究势能面的相交规则. 先讨论双原子分子的势能曲线,此时,我们有如下的相交规则: 对称性(包括自旋和空间)相同的态的势能曲线不能相交,对称性不同的态 的势能曲线可以相交. 简要解释一下上述规则. 我们知道,势能曲线是电子哈密顿量的本征能量曲 线,相应的本征函数应具有两种对称性,一是自旋对称性,即电子波函数应该是自旋算符的本征函数,总自旋算符有确定值S ,通常用(21)S +表示电子波函数的自旋多重度. 此外,电子波函数还应具有分子所属点群的对称性,即它应当是分子所属点群不可约表示的基. 上述规则意味着,当两个电子态波函数具有相同的自旋多重度并属于分子点群同一不可约表示时,相应的势能曲线不能相交,称之为避免交叉. 现在来证明这一规则. 双原子分子具有h D ∞(同核)或h C ∞(异核)对称性. 设1 (21)()1(,)S R q +λΨ和2(21) ()2(,)S μR q +ψ为某一双原子分子的两个正交归一化 的非简并电子态波函数,分别为点群的λ和μ不可约表示的基,自旋多重度分别为1(21)S +和2(21)S +,相应的势能曲线分别记作)(1R E 和)(2R E . 如图(1.14) 所示。 图1.14 势能曲线相交 假定两曲线相交于c R 点,即)()(21c c R E R E =,并设0R 为c R 附近的一点,记 )(00R H H =, )(0101R E E =, )() (,)1(2S 10110R q +λ=ψψ, )(0202R E E =,)()(,)2(2S 10220R q +μ=ψψ, 则有: 0000111H E =ψψ (1.7.1) 00 00222H E =ψψ (1.7.2) 假定核间距有一微小变化R δ,R R R δ+=0,这时体系的Hamilton 算符相应变为 00R R H H H R H V R δ=???=+=+ ? ??? (1.7.3) 式中, 0R R H V R R δ=??? = ? ??? 相应的Schr?dinger 方程为 );();(q R E q R H ψ=ψ (1.7.4) 势能曲线是R 的连续函数,由于)()(21c c R E R E =,在c R 附近的任何点R ,都应有)()(21R E R E ≈,因此可按简并态微扰方法求解方程(1.7.4),于是有 022011);(ψ+ψ=ψc c q R (1.7.5) 代入(1.7.4)式,分别左乘*01ψ和* 02ψ,并对电子坐标积分,可得久期方程为: 02221 1211=--E H V V E H (1.7.6) 式中,0000ii i ii i i i H E V E V =+=+ψψ,0o ij i j V V =ψψ. 假定不考虑自旋-轨道 耦合(体系中不含重原子),则01ψ和02ψ可以选为实函数,这时12V 和21V 为实数,并有1221V V =. 由(1.7.6)式可解得 ()1 22 2112211221211()422E H H H H V ±??=+±-+? ? (1.7.7) 相应的波函数分别为 分子反应动力学(势能面)的基本概念 搜索的内容:各种概念介绍 分子反应动力学:分为:宏观反应动力学(Macroscopic Kinetics) 微观反应动力学(Microscopic Kinetics)即为分子反应动力学(Molecular Reaction Dynamics)。 (不同定义表述) 1.在原子、分子的层次上研究化学反应微观动态和机理的一门科学,它所研究的基元反应和基元化学物理过程能够使人们了解化学反应的机理。 2.应用现代物理化学的先进分析方法,在原子、分子的层次上研究不同状态下和不同分子体系中单分子的基元化学反应的动态结构,反应过程和反应机理。(张爱丽) 3.分子反应动力学是现代物理与化学之间的一门边缘学科,是化学物理学科的一个重要分支。它深入到分子或原子层次来研究化学反应的微观动态和机理。分子反应动力学的研究主要包括: 1)构建反应体系的势能面; 2)计算该体系的微观动力学参量(如截面),这些参量是反应物的初态及产物终态的函数;3)通过积分截面得到宏观动力学参量(速率常数) 注: 基元反应:在反应中一步直接转化为产物的反应(又称简单反应)。基元反应本身是指没有中间产物,一步完成的反应。目前验证基元反应最科学的方法包括量子化学的模拟计算和以飞秒激光为代表的分子动力学手段。通过计算机模拟反应过程可以得到一个反应的模拟过程,数据时很好的预测手段。通过飞秒激 光得到反应过程中各种物质的光谱变化,可以推断反应过程中到底什么物质或者是物质的什么状态发生反应,从而最终确定反应的过程。(张爱丽) 势能面的构建 势能面的意义: 基于电子运动和核运动可分离假定的势能面概念是现代化学物理学最重要的思想之一。 从动力学理论计算的角度来讲,势能面是最基本也是非常重要的一个因素,势能面的准确程度对动力学计算的结果有直接影响。势能面的形状反映出整个化学反应过程的全貌以及反应的始终态、中间体和过渡态的基本态势。在势能面上连接这些态的一条最容易实现的途径就是整个化学反应的路径。势能面上反应体系反应坐标的各种物理化学性质的变化,提供了反应历程的详尽信息。势能面提供了反应过程的舞台,它包含了整个反应过程的信息库。 获得正确的势能面是从理论上研究化学反应的首要任务。 势能面的构建理论: 目前势能面的来源主要有两种:一种是在从头算基础上的数值拟合,一种是利用半经验表达形式确定参数。第一种方法:原则上是可以精确描述化学反应。具体方法是:借助从头算得到的一些分立几何构型点的能量,然后借助这些分立的能量点做势能面拟合。 非绝热效应:电子的非绝热过程普遍存在于光化学反应、激发态物种之间的碰撞、燃烧反应、异质溶解过程和电荷转移过程之中。目前除了对三原子体系的非绝热反应过程的研究之外,已经拓展到了四原子以及更多原子的反应体系。(张爱丽) 乙烷及乙烷取代物绕C-C键旋转势能面扫描 方法步骤:(内坐标法scan): 1.同上用gaussianview做输入文件:分别构建乙烷,乙醇,乙二醇分子,保存为11.gjf 1 2.gjf 1 3.gjfG03输入文件格式。 2.修改gaussianview生成的内坐标:分别用gaussianview打开11.gjf 12.gjf 1 3.gjf并显示原子编号,根据图像对应坐标修改,使其只有一个二面角的改变可引起乙烷两个甲基相对旋转。如下所示乙烷; 0 1 C H 1 B1 H 1 B2 2 A1 H 1 B3 3 A2 2 D1 C 1 B4 4 A3 3 D2 H 5 B5 1 A4 4 D3 H 5 B6 1 A5 4(6)D4 H 5 B7 1 A6 4(7 )D5 B1 1.07000000 B2 1.07000000 B3 1.07000000 B4 1.54000000 B5 1.07000000 B6 1.07000000 B7 1.07000000 A1 109.47120255 A2 109.47121829 A3 109.47121829 A4 109.47120255 A5 109.47120255 A6 109.47123134 D1 -120.00000060 D2 120.00003407 D3 179.98890060 D4 59.88891539 (115.) D5 179.88890799 (115.) 然后进行正常优化和频率分析(#hf/3-21g opt freq )。 3.建立扫描文件:同前判断11.OUT .12.OUT 13.OUT优化结果,2个normal4个yes,无虚频。然后分别在11.OUT .12.OUT 13.OUT中找到Final structure in terms of initial Z-matrix:把对应坐标粘贴建立扫描文件11scan.gjf.12scan.gjf.13scan.gjf。 4. 扫描结果;用Summary of the potential surface scan处所对应的SCF值和步数origin6.0作图。 搜索的内容:各种概念介绍 分子反应动力学:分为:宏观反应动力学(Macroscopic Kinetics) 微观反应动力学(Microscopic Kinetics)即为分子反应动力学(Molecular Reaction Dynamics)。 (不同定义表述) 1.在原子、分子的层次上研究化学反应微观动态和机理的一门科学,它所研究的基元反应和基元化学物理过程能够使人们了解化学反应的机理。 2.应用现代物理化学的先进分析方法,在原子、分子的层次上研究不同状态下和不同分子体系中单分子的基元化学反应的动态结构,反应过程和反应机理。(张爱丽) 3.分子反应动力学是现代物理与化学之间的一门边缘学科,是化学物理学科的一个重要分支。它深入到分子或原子层次来研究化学反应的微观动态和机理。 分子反应动力学的研究主要包括: 1)构建反应体系的势能面;2)计算该体系的微观动力学参量(如截面),这些参量是反应物的初态及产物终态的函数;3)通过积分截面得到宏观动力学参量(速率常数) 注: 基元反应:在反应中一步直接转化为产物的反应(又称简单反应)。基元反应本身是指没有中间产物,一步完成的反应。目前验证基元反应最科学的方法包括量子化学的模拟计算和以飞秒激光为代表的分子动力学手段。通过计算机模拟反应过程可以得到一个反应的模拟过程,数据时很好的预测手段。通过飞秒激光得到反应过程中各种物质的光谱变化,可以推断反应过程中到底什么物质或者是物质的什么状态发生反应,从而最终确定反应的过程。(张爱丽) 势能面的构建 势能面的意义: 基于电子运动和核运动可分离假定的势能面概念是现代化学物理学最重要的思想之一。 从动力学理论计算的角度来讲,势能面是最基本也是非常重要的一个因素,势能面的准确程度对动力学计算的结果有直接影响。势能面的形状反映出整个化学反应过程的全貌以及反应的始终态、中间体和过渡态的基本态势。在势能面上连接这些态的一条最容易实现的途径就是整个化学反应的路径。势能面上反应体系反应坐标的各种物理化学性质的变化,提供了反应历程的详尽信息。势能面提供了反应过程的舞台,它包含了整个反应过程的信息库。 获得正确的势能面是从理论上研究化学反应的首要任务。 势能面的构建理论: 目前势能面的来源主要有两种:一种是在从头算基础上的数值拟合,一种是利用半经验表达形式确定参数。第一种方法:原则上是可以精确描述化学反应。具体方法是:借助从头算得 到的一些分立几何构型点的能量,然后借助这些分立的能量点做势能面拟合。 非绝热效应:电子的非绝热过程普遍存在于光化学反应、激发态物种之间的碰撞、燃烧反应、异质溶解过程和电荷转移过程之中。目前除了对三原子体系的非绝热反应过程的研究之外,已经拓展到了四原子以及更多原子的反应体系。(张爱丽) 光化学反应:一个原子、分子、自由基或离子吸收一个光子所引发的化学反应。光化学反应是由物质的分子吸收光子后所引发的反应。分子吸收光子后,内部的电子发生能级跃迁,形成不稳定的激发态,然后进一步发生离解或其它反应。(张爱丽) 电子的绝热过程:B-O-A,又称为绝热近似(定核近似)——就是在研究分子时,将电子的运动 势能面与构型优化 α = 30o 内坐标:坐标自由度数目为3N-6 A. 极小点 势能面中,所有的“山谷”为极小点,对这样的点,向任何方向在势能面上移动—轻微改变结构,将引起势能升高。 极小点可以是区域极小点(在有限区域内的),也可以是全局(整个势能面上)极小点。 极小点对应于体系的平衡结构,对单一分子不同的极小点对应于不同的构象或结构异构体。对于反应体系极小点对应于反应物、产物,反应中间物等。 考虑到量子化学是对静态的体系进行研究,极小点是体系真实性质的代表点,因此是研究的重点。 这些极小点从数学意义上来讲是势能对坐标的一阶导数为零,而二阶导数为正(Hessian 矩阵本征值为正),因此可以用数学的方法搜索(如优化等)。需要注意的是,一般优化方法仅可找到初始构型附近的极小点,因此优化的初始构型非常重要。 对于极小点,如果偏离位置则受到相反方向的力,因此可以计算出振动频率。振动频率对应分子光谱(IR, Ramman) 一般优化过程为节省时间,其Hessian矩阵本征值采用的是估算,因此要严格确定所优化的结果是否是真正的极小点需要作频率分析,所计算出的频率应均为正。如出现负值,一般可能是对称性限制引起的。 B. 鞍点 势能面上的另一类重要的不动点为鞍点(更严格应称一阶鞍点),这些鞍点是连接两个极小点中间最底的“山口”,对应于化学反应体系的过渡态(或构型变化中的中间态)。 从数学意义上,在鞍点处势能对坐标的一阶导数为零,而Hessian矩阵本征值只有一个负值。鞍点是在其中一个方向上具有极大,而其它方向均为极小。鞍点是由于其形状如马鞍而得名。同极小点类似,严格的鞍点需要进行频率分析验证,必须有且只有一个虚频率(频率为负)。C. 最小能量途径(minimum energy path) 最小能量途径(MEP)是连接势能面上两个极 小点之间最低的能量途径,MEP也称内禀反应坐标(intrinsic reaction coordinate —IRC)。MEP形象 形容是从鞍点放置一个球,球在势能面上自然滚落,并且起速度在每经过的点都得到充分的阻尼,最后落到极小点所经过的路径。当势能面使用质量权重坐标时,MEP为最快下降途径(steepest gradient)。势能面相交规则
ADF教程:如何进行势能面扫描
势能面相交规则
分子反应动力学(势能面)的基本概念讲课讲稿
势能面扫描
分子反应动力学(势能面)的基本概念
3势能面与构型优化