包装材料质量标准
一、目的:
明确产品所用包材质量要求,严格按本标准采购、检验、生产,保证产品的质量。 二、范围:
适用于本企业的75%单方乙醇消毒液的检验及质量控制。 三、职责:
1、 供应部:严格按本标准要求,从定点供应商采购;
2、 仓库:严格按本标准接收、贮存原料;
3、 品质部:检验员严格按本质量标准检验,认真、及时、准确地填写检验记录,化验室 负责人监督检查检验员执行本标准,QA 按本标准放行前审核;
4、 生产部:严格按本质量标准使用包装材料。 四、内容:
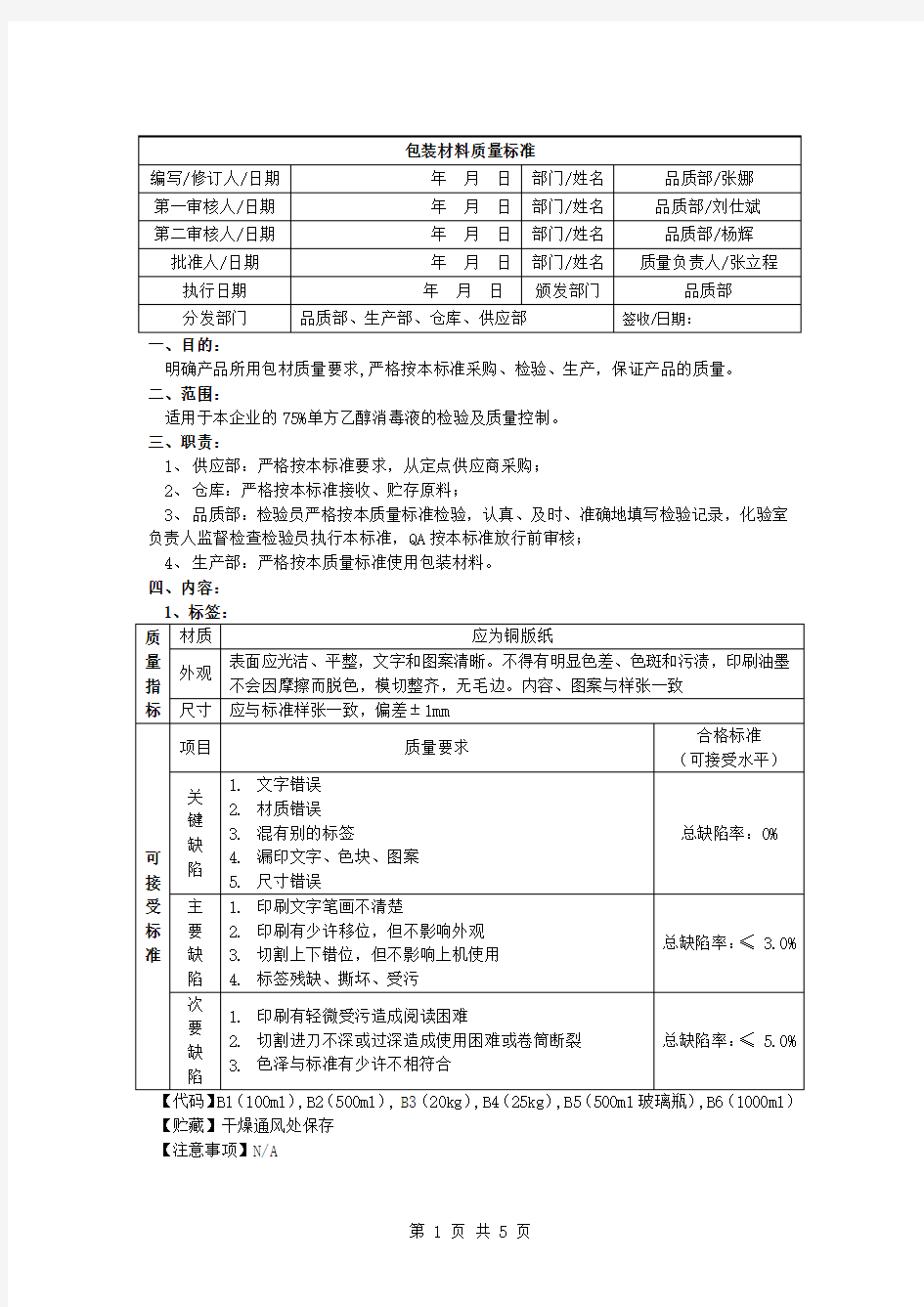
【代码】B1(100ml ),B2(500ml )
, B3(20kg ),B4(25kg ),B5(500ml 玻璃瓶),B6(1000ml )
【贮藏】干燥通风处保存 【注意事项】N/A
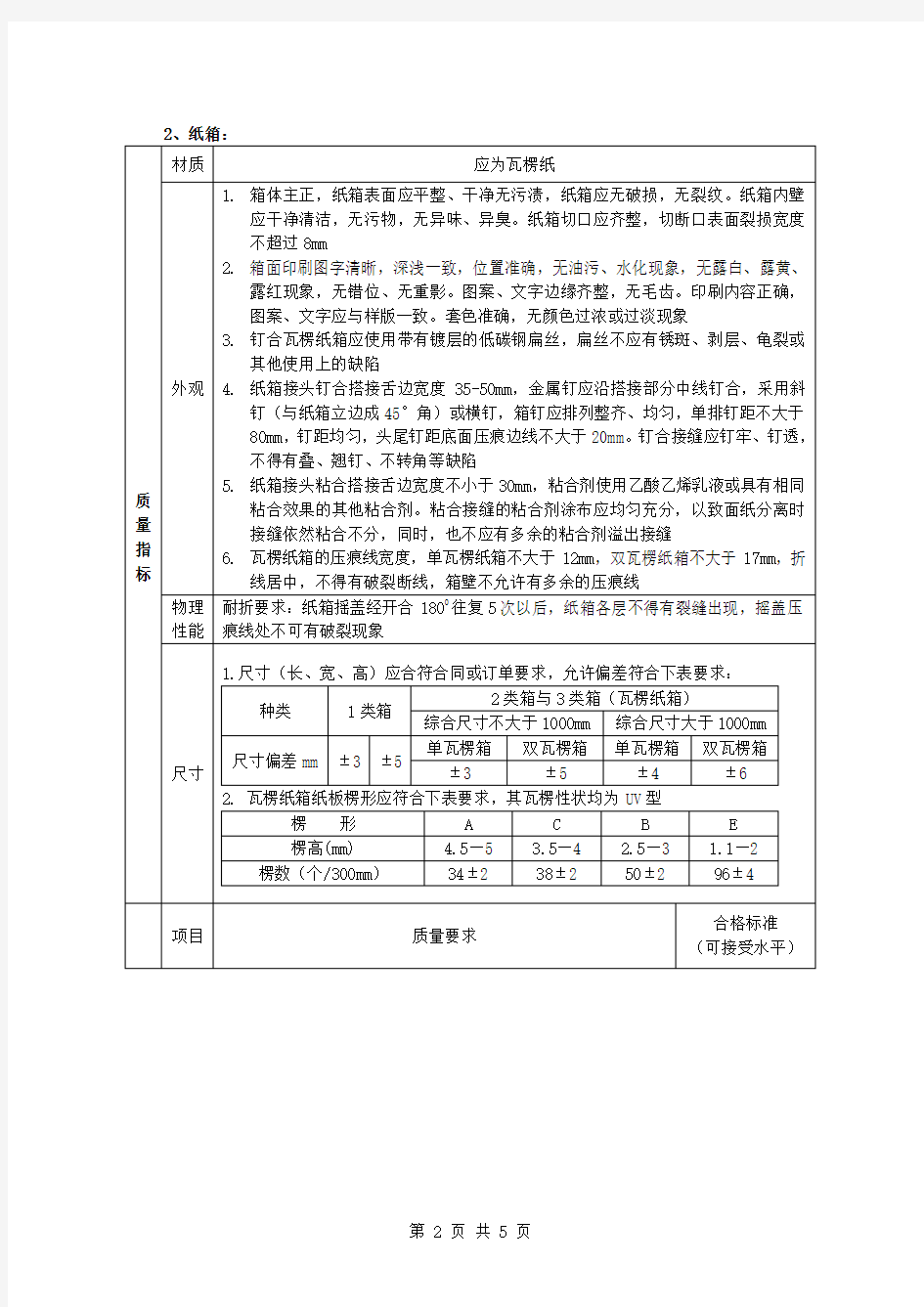
2、纸箱:
【代码】Bh-1(100ml),Bh-2(500ml), Bh-5(500ml玻璃瓶),Bh-6(1000ml)【贮藏】干燥通风处保存
【注意事项】N/A
【贮藏】干燥通风处保存
【注意事项】生产上遇急需使用包材时,可在微生物结果出来前使用,但微生物检测结果一旦不合格则应进行返工
【贮藏】干燥通风处保存
【注意事项】生产上遇急需使用包材时,可在微生物结果出来前使用,但微生物检测结果一旦不合格则应进行返工
【代码】BP-5(500ml玻璃瓶)
【贮藏】干燥通风处保存
【注意事项】生产上遇急需使用包材时,可在微生物结果出来前使用,但微生物检测结果一旦不合格则应进行返工
五、参考文献:
《药品生产质量管理规范》
六、相关文件:
包装材料检验操作规程
七、相关记录:
N/A
八、变更记录及原因:
种进口中药材质量标准原件
43种进口药材质量标准的通知关于颁布儿茶等 [2004]144号国食药监注省、自治区、直辖市食品药品监督管理局(药品监督管理局),各口岸食品药品监督管局(药品监督管理局),各口岸药品检验所:为加强进口药材的监督管理,我局修(制)订了儿茶、方儿茶、西洋参、高丽红参、红花、牛黄、羚羊角、泰国安息香、苏合香、乳香、没药、血竭、藤黄、沉香、檀香、香、母丁香、小茴香、荜茇、广天仙子、豆蔻、槟榔、肉豆蔻、大腹皮、大风子、西青、诃子、胖大海、芦荟、猴枣、弗朗鼠李皮、胡黄连、肉桂、番泻叶、马钱子、玳瑁、种进口药材的质量标准(见43决明、天竺黄、穿山甲、海狗肾、海马、哈蚧、海龙等件),现予颁布。上述质量标准自颁布之日起执行。国家食品药品监督管理局 四年五月八日二○○ 中药材质量标准 中文名儿茶 汉语拼音Ercha 英文名CA TECHU 来源儿茶为豆科植物儿茶Acacia catechu(.)Willd.的去皮后的枝干的干燥煎膏。冬季采收枝、干,除去外皮,砍成大块,加煎煮,浓缩,干燥。 性状本品呈方形或不规则块状,大小不一。表面棕褐色或黑褐色光滑而略有光泽。质硬,易碎,断面不整齐,具光泽,有细. 孔,遇潮有黏性。无臭,味涩、苦,略回甜。 鉴别(1)本品粉末棕褐色。可见针状结晶及黄褐色块状物。 (2)取火柴杆浸于本品水溶液中,使轻微着色,待干燥后,浸入盐酸中立即取出,置火焰附近烘烤,杆上即显深红色。(3)取本品粉末约,加乙醚30ml,超声处理10分钟,过,滤液蒸干,残渣用甲醇5ml 使溶解,作为供试品溶液。另取儿茶素和表儿茶素照品,加甲醇制成每1ml各含的混合溶液,作为对照品溶液。照薄层色谱法(中国药典B)2000年版一部附录Ⅵ 试验,吸取供试品溶液5?l,对照品溶液2?l,分别点于同一维素预制板上,以正丁醇—醋酸—水(3:2:1,上层)(实验室室内相对湿度要求在4 以下)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,热风吹至斑点显色清晰供试品色谱中,在与对照品色谱相应位置上,显相同的红棕色斑点。 检查淀粉显微镜下观察,本品粉末以水装置,不得有淀粉粒。水分照水分测定法(中国药典2000年版一部附录%。H第一法)测定,不得过总灰分不得过%(中国药典2000年版一部附录IX K) 浸出物含量测定照高效液相色谱法(中国药典2000年版一部附录ⅥD测定。色谱条件与系统适用性试验用十八烷基硅烷键柠檬酸硅胶为填充剂;L—N,N-二甲基甲酰胺—四氢呋喃(45:8:2V/V)为流动相;检测波长为280nm;柱温:。℃。理论板数按儿茶素峰计算应不得低于300035对照品溶液的制备精密称取儿茶素、表儿茶素对照品11量,加甲醇—水(:)制成每含儿茶素、表儿茶素的溶液,即得。1ml供试品溶液的制备取本品粉末,精密称定,置50ml量:1瓶中,加甲醇—水(1)40ml,超声处理20分钟,并以甲醇—水(1:1)稀释至?m度,摇匀,用微孔滤膜()滤过,即得。分别精密吸取上述两种对照品溶液与供试品溶测定法 ,注入液相色谱仪,测lμ5各. 定,即得。 本品含儿茶素(C15H14O6)和表儿茶素(C15H14O6)。总量不得少于%
固体药用聚烯烃塑料瓶质量标准及检验规程
目的:明确固体药用聚烯烃塑料瓶的质量标准和规范药用聚烯烃塑料瓶的检验。 适用范围:适用于包装非芳酸性、非油酸性、非挥发性及易氧化的固体药品(片剂、胶囊、制剂)的塑料瓶。 责任者:化验员。 引用标准:YY0057-91 GB2828 1、材料 高密度聚乙烯树脂或聚丙烯树脂为主要原料。 2、技术要求 2.1 药用塑料瓶的外观质量:应具有均匀一致的乳白色泽,不得有明显的色差。瓶的表面应光洁、平整、不允许有变形和明显的擦痕。不允许有砂痕、油污、气泡。瓶口应平整、光滑。 2.2 物理性能应符合表1规定: 表1 2.3 化学性能符合表2规定: 2.4 菌检验应符合以下规定:小于108ml的塑料细菌总数不超过1500个/瓶,霉菌
总数不得超过150个/瓶;100ml至250ml的塑料瓶细菌总数不超过3000个/瓶,霉菌总数不得超过300个/瓶;大于250ml的塑料瓶细菌总数不得超过3500个/瓶,霉菌总数不得超过350个/瓶。所有规定的塑料瓶大肠肝菌均不得检出。 2.5 异常毒性:无异常毒性 3、试验方法 3.1 外观 在自然光线明亮处目测检验。 3.2 密封性试验 每个瓶装进一定量的玻璃球,紧盖后(带有螺旋盖的试瓶用测力扳手将瓶与盖旋紧,扭力见表3)置于带有抽气装置的容器内,用水浸没,抽真空到26.67kpa维持2min,瓶内不得有进水或冒泡现象。 表3 3.3 振荡试验 每个瓶装入酸性水为标示剂、紧盖后(带有螺旋盖的试瓶用测力扳手将盖与瓶旋紧,扭力见表3)用溴酚蓝试纸(将滤纸浸入稀释5倍的溴酚蓝试液,浸透后取出干燥)紧包瓶的颈部,置振荡器(振荡器频率每分钟200次±5%)振荡30min后,溴酚蓝试纸不变色为合格。 3.4 水蒸气渗透量试验 每个试瓶用绸布擦净,将瓶盖连续开、关30次后,在试瓶内加入无水氯化钙干燥剂(除去过4目筛过细粉,置110℃干燥1h),20ml或20ml以上的试瓶,加干燥剂量为13mm高,小于20ml的试瓶,加入干燥剂量为容积2/3;如试瓶高度超过63mm,加入干燥剂量为50mm高,立即将盖盖紧。另取两个试瓶装入与干燥剂相等量的玻璃小球,作对照用。试瓶紧盖后分别称定重量,然后将试瓶量相对湿度为100%,温度达25±2℃,放置72h,取出,室温放置45min,分别称量。按式(1)计算水蒸气渗透量。 1000 水蒸气渗透量(mg/24h.L)=————[(T 2-T 1 )-(C 2 -C 1 )] (1) 3V 式中:V——试瓶的容积,ml; T 1 ——试瓶试验前的重量,mg;
药用包装材料质量标准ISO15378(中文)
精心整理 ISO15378:2006标准的中文版内容 国际标准化组织/第76技术委员会(IS0/TC76)于2003年制定了1S015378国际标准草案(Draftofinterrfationalstandard ,D1S),标题是:《药品初包装材料ISO9001:2008应用的专用要求,包含生产质量管理规范(GMP)》。2006年,形成了借鉴ISO9001:2008质量体系的《药用包装材料质量标准ISO15378:2006》初稿。这个国际标准的制定说明了国际社会对药包材生产企业实施质量管理的重视。此处择其(一) 0.1领会GMP 采用0.2(1)理解并满足要求; (2)需要从增值的角度考虑过程; (3)获得过程业绩和有效性的结果; (4)基于客观的测量,持续改进过程。 ISO15378引用了以过程为基础的QMS 模式图,展示了有关的过程联系(可参见ISO9001:2008)。 此外,称之为“PDCA ”的(Plan 计划,D0执行,Check 检查,Action 处理)的方法可
适用于所有过程。 0.3与ISO9004的关系 ISO9001与ISO9004为一对协调一致的QMS标准,它们相对补充,但也可单独使用。 ISO9001规定了QMS要求,可供组织内部使用,也可用于认证或合同目的。在满足顾客要求方面,ISO_9001,所关注的是QMS的有效性。 与ISO9001相比,ISO9004为QMS更宽范围的目标提供了指南。除了有效性,该标准还特别关注持续改进组织的总体业绩与效率。对于最高管理者希望通过追求业 0.4 这些 )、OHSMS( (二) 1.范围 1.1 认为该项要求“适当”:初包装材料满足所规定的要求;和(或)组织实施纠正措施。 1.2应用 1S015378是一个药品初包装材料的应用标准。该标准也适用于第三方对此类产品的认证。 2.规范性引用文件 ISO9000:2008质量管理体系基础和术语 ISO9001:2008质量管理体系要求
中药质量标准提高原则
提高中成药质量标准指导原则和技术要求 一、概述 药品标准是衡量药品质量的尺度和准则。按照《药品管理法》的规定,“药品必须符合国家药品标准”,“国务院药品监督管理部门颁布的《中华人民共和国药典》和药品标准为国家药品标准”。国家药品标准是国家对药品的质量和检验方法所做的技术规定,是在正常的原辅料与正常的生产条件下生产的药品质量是否符合要求的判断标准,是药品生产、销售、使用和检验部门共同遵守的法定依据。 随着科技的进步与发展,药品质量控制与检验技术也在不断进步与发展。为保障公众安全有效用药,积极应用现代药品质量控制与检验技术,国家食品药品监管局作出了“提高国家药品标准行动计划”的战略决策,并在国家药典委员会的具体组织下全面开展,中成药成为首先实施的药品类别。 广东省是中药大省,中成药品种数量在全国名列前茅。贯彻实施国家药品标准提高行动计划,规范广东省提高中成药质量标准工作,对保障公众用药安全有效,提高中成药质量控制和检验技术水平,提高广东中成药产业竞争力具有重要意义。根据国家食品药品监管局和国家药典委员会的有关规定,特制定《广东省提高中成药质量标准指导原则和技术要求》,供广东省中成药研究单位、生产企业和药品检验机构在提高中成药质量标准工作中使用。 二、指导原则 “科学、规范、实用”是国家药品标准制定的总的指导原则,是国家药品标准的灵魂和 精神所在。广东省提高中成药质量标准的指导原则是在认真贯彻“科学、规范、实用”这一 总的指导原则的基础上,根据广东中成药产业的实际,经过有关专家广泛深入讨论后形成的。 (一)质量标准的可控性原则 “质量可控”是药品标准的目标性原则。为实现“质量可控”,药品标准的建立应充分 考虑药品在来源、生产、流通以及使用等各个环节可能影响药品质量的因素,有针对性的确 定标准制订或提高的内容,建立相应的检测方法。药品的质量标准应能反映药品的内在质量, 必须能够有效地控制药品的质量,以确保药品的安全和有效。 (二)检测方法的科学性原则 “准确灵敏”是检测方法选用的科学性原则。检测方法在可控的基础上应尽可能体现与 真实值接近的准确性,最大限度减少各种偏差,同时体现该检测方法对被测药品的专属性。 检测方法的建立应包括: 1.分析方法的选择 目前各种色谱方法、光谱方法和经典测定方法广泛应用于中药材及其制剂的检测。质量 标准中分析方法的选择应与被测成分的性质相适应,与被测成分的含量限度相适应,与应用 的具体要求相适应,并能有效排除干扰成分的影响。 2.分析方法的设计 中药有效成分或指标成分的检测方法已有大量的文献报道。检测方法的研究首先应查阅
GMP包装材料质量标准(1)
一、目的:制定通用纸箱的质量标准,规范公司通用纸箱的采购、检验、使用。 二、标准依据:《中华人民共和国国家标准》GB6543-86。 三、适用范围:适用于公司通用纸箱的采购、检验。 四、责任者:质保部全体人员、仓库管理员、采购员。 五、内容: 通用纸箱 B-01、B-02、B-03、B-45 【材质】本品材质为卡面纸、瓦楞纸板。 【外观】合拢摇盖,离缝重叠不得大于3mm。 箱体方正,表面没有明显的损坏和污迹,切断口表面裂损宽度不超过8mm。 箱面印刷文字、图案清晰深浅一致,位置准确,色泽鲜艳,颜色均匀。 纸箱接头钉合搭接舌边宽度35~50mm,金属钉应沿搭接部分中线钉合,箱钉应排列整齐、均匀,单排钉距不大于80mm,钉距均匀,头尾钉距底面压痕边线不超过20mm。 纸箱接头粘合搭接舌边宽度不小于30mm,粘合接缝的粘合剂涂布应均匀充分。 瓦楞纸箱的压痕线宽度,单瓦楞纸箱不大于12mm,双瓦楞纸箱不大于17mm,折
线居中,不得有破裂断线,箱壁没有多余的压痕线。 瓦楞纸箱摇盖经开、合180o往复5次,不得有裂缝。 粘合纸箱用乙酸乙烯乳液或具有同等效果的其它黏合剂。 纸箱附件纸箱附件应与纸箱配套。 规格箱子的基本尺寸符合下表要求: 允许误差±5mm、±5g
一、目的:制定塑料瓶的质量标准,规范公司塑料瓶的采购、检验、使用。 二、标准依据:《国家药品包装容器(材料)》标准、《中华人民共和国国家标准》 GB5009·60—85。 三、适用范围:适用于公司口服液药用瓶、液体消毒剂和杀虫剂用塑料瓶的采购、检验。 四、责任者:质保部全体人员、采购员、仓库管理员。 五、内容: 塑料瓶 【材质】口服液药用瓶的材质为聚对苯二甲酸乙二醇酯塑料;液体消毒剂和杀虫剂用瓶材质为聚丙烯塑料。 【外观】表面应清洁、光滑、无破损。 无污迹、卫生、洁净。 瓶盖应清洁、无破损、硬度好,与塑料瓶结合良好,气密性好。 规格尺寸:允许误差±2mm、±0.5g 口服液药用瓶应按下述方法进行微生物限度检测。 【微生物限度】按照微生物限度检查法检测,将口服液药用瓶用50ml0.9%灭菌氯化钠液冲洗内壁,作为供试液。取适宜的连续的2-3个稀释级的供试液1:10、1:100、1:1000的稀释液各1ml,置直径约90mm的平皿中,再注入约45℃的培养基约15ml,混匀,待凝固后,倒置培养,每稀释级作2个平皿。
新的药用辅料注册申报资料要求
一、新的药用辅料注册申报资料要求(一)综述资料1、药用辅料名称(包括正式品名、化学名、英文名称、汉语拼音等)以及命名的依据。2、证明性文件:(1)申请人合法登记证明文件、《药品生产许可证》复印件;(2)药用辅料或者使用的处方、工艺等专利情况及其权属状态说明,以及对他人的专利不构成侵权的保证书。3、立题目的与依据。包括国内外有关该品研发、上市销售及相关文献资料、生产及在制剂中应用情况综述。4、对主要研究结果的总结及评价。包括申请人对主要研究结果进行的总结,并从安全性、有效性、质量可控性等方面对所申报的品种进行综合评价。5、说明书样稿、起草说明及最新参考文献。包括药用辅料名称、化学结构式或分子式、用途、注意事项、包装(规格、含量)等,须注明有效期,并应明显标注“药用辅料”的字样。6、包装、标签设计样稿。二)药学研究资料7、药学研究资料综述。包括合成工艺、处方筛选、结构确证、质量研究和质量标准制定、稳定性研究等的试验和国内外文献资料的综述。 8、生产工艺的研究资料及文献资料。包括制备的工艺流程和化学反应式、起始原料和有机溶媒、反应条件(温度、压力、时间、催化剂等)和操作步骤、精制方法、主要理化常数及阶段性的数据累计结果等,并注明投料量和收得率以及工艺过程中可能产生或引入的杂质或其他中间产物,提供所用化学 原料的规格标准,动、植、矿物原料的来源、学名。凡制备工艺与主要参考文献不同者,应提出修改的依据。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。包括理化常数、纯度检验、含量测定及方法学验证及阶段性的数据积累结果等。 11、与药物相关的配伍试验资料及文献资料。 12、标准草案及起草说明,并提供标准品或者对照品。质量标准应当符合《中国药典》现行版的格式,并使用其术语和计量单位。所用试药、试液、缓冲液、滴定液等,应当采用现行版《中国药典》收载的品种及浓度,有不同的,应详细说明。提供的标准品或对照品应另附资料,说明其来源、理化常数、纯度、含量及其测定方法和数据。 标准起草说明应当包括标准中控制项目的选定、方法选择检查及纯度和限度范围等的制定依据 13、连续3批样品的检验报告书。指申报样品的自检报告。14、稳定性研究的试验资料及文献资料。包括采用直接接触药用辅料的包装材料和容器共同进行的稳定性试验。 15、直接接触药用辅料的包装材料和容器的选择依据及质量标准。(三)药理毒理研究资料16、药理毒理研究资料综述。17、对拟应用药物的药效学影响试验资料及文献资料。18、一般药理研究的试验资料及文献资料。19、急性毒性试验资料及文献资料。20、长期毒性试验资料及文献资料。21、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等主要与局部、全身给药相关的特殊安全性试验研究和文献资料。 22、致突变试验资料及文献资料。 23、生殖毒性试验资料及文献资料。 24、致癌试验资料及文献资料。(四)临床研究资料 25、国内外相关的临床研究资26、临床研究计划及研究方案。 27、临床研究者手册。 28、知情同意书样稿、伦理委员会批准件。 29、临床研究报告。 三、已有国家标准的药用辅料注册申报资料要求(一)综述资料 1、药用辅料名称(包括正式品名、化学名、英文名称、汉语拼音等)以及命名的依据。 2、证明性文件: (1)申请人合法登记证明文件、《药品生产许可证》复印件;
中药材质量标准-大腹皮
中药材质量标准 中文名大腹皮 汉语拼音Dafupi 英文名PERICARPIUM ARECAE 来源本品为棕榈种植物槟榔Areca catechu L.经加工除去外果皮的干燥成熟果皮。 性状本品略呈椭圆形,均纵剖成瓣,似瓢状。中果皮黄白色或淡棕色,疏松柔韧,纤维性棕毛状。内果皮硬壳状,黄棕色至深棕色,内表面光滑,有时纵向破裂。无臭,无味。 鉴别(1)本品粉末黄白色或黄棕色,中果皮纤维成束,细长,直径8?15 口,微木化,纹孔明显,周围细胞中含有圆簇状硅质块,直径约8g。内果皮细胞呈不规则多角形、类 圆形或椭圆形,直径48?88 g,纹孔明显。 (2)取本品粉末2g,加盐酸-水(1: 100)50ml,加热煮沸,并保持微沸 5分钟,冷却,滤过,滤液弃去,药渣加乙醇50ml,水浴加热回流30分钟,滤过,滤液 蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液。另取大腹皮对照药材2g,同法制 成对照药材溶液。照薄层色谱法(中国药典2000 年版一部附录W B)试验,吸取上述 两种溶液各2』,分别点于同一硅胶 G薄层板上,以环已烷醋酸乙酯浓氨试液(10 : 5: 0.1)为展开剂,展开,取出,晾 干,喷以5%磷钼酸乙醇溶液,105C烘约10分钟。供试晶色谱中,在与对照药材色谱 相应的位置上,显相同颜色的斑点。 检查取本品不少于5Kg,检查杂质、霉变。 杂质,霉变取本品(不少于5Kg)检查,杂质(指残存的槟榔及其他有机、无机杂质,包括逐个抖下的碎末和泥砂等)不得过4%;霉变(指 发霉变黑,其黑色由外表面深入到内部且占整个体积的1/3以上者)不得 过5%。 干燥失重不得过12.0%。取本品约5g,精密称定,小心铺在直径约9cm的平皿或培养皿内,于105C干燥5小时,移置干燥器中,冷却30 分钟,迅速精密称定重量, 计算,即得。 浸出物取本品粉末4g,称定重量,置150ml锥形瓶中,精密加入乙醇50ml,塞紧,称定重量,静置1小时后,连接回流冷凝管,加热至沸腾,并保持微沸1小时。放冷,取下 锥形瓶,密塞,称定重量,用乙醇补足减失的重量,摇匀,用干燥滤器滤过。精密量取滤 液25ml,置已干燥至恒重的蒸发 皿中,在水浴上蒸干后,于105C干燥3小时,移置干燥器中,冷却30分钟,迅速精 密称定重量,计算,即得。 本品以干燥品计算,不得少于2.0%. 含量测定 炮制除去杂质,洗净,干燥。
药用塑料瓶的吹塑成型工艺
药用塑料瓶的吹塑成型工艺 一、简介 世界上“注-吹”成型工艺方法源于二十世纪五十年代初。国外聚烯烃(hdpe,pp)药用塑料瓶的应用早于七十年代。相关设备研究制造厂家有美国wheaton、jomar,德国battenfeld、bekum,日本的asb、青木固,意大利的uniloy milacron公司,主要机型均为一步法三工位,美国、日本采用垂直螺杆结构,德国、意大利采用卧式螺杆结构。 国内第一批系统引进“注-吹”流水线并生产药品包装塑料容器的企业有天津力生制药厂(1984年)和上海大明玻璃厂(1985年)。并为我国消化吸收全面推广聚烯烃药用塑料瓶做了大量基础性工作。 江苏维达机械有限公司自1989年首家研制“三工位”一步法“注-吹”成型机,于1991年首批投放市场,经过十多年的努力,取得了长足进步。随着我国医药工业的发展及广大用户的支持,近年来以每年100条生产线的规模高速发展,使得中国药品包装容器(尤其是固体药物片剂、胶囊包装)整体质量水平得到了较大幅度的提升。 二、药用塑料瓶常用生产工艺与原料选择 1、生产工艺 (1)“挤吹”extrusion-blow moulding又称中空挤出吹塑。挤出机连续挤出空心管,用剪刀(人工)或切割装置(自动)切成小段后移到挤吹模具内吹制成型。 优点:设备简单、投资小,成本价格低。 缺点:瓶口不平,密封很差。原料通常选用ldpe,阻透性能远低于hdpe/pp,装药保质贮存期短。 (2)二步法“注-吹”two steps injection-blow moulding。“注射、吹塑”由独立的两台机器分开进行,俗称“二步法”。第一步:由一台普通注塑机注射成型管坯,管坯的瓶头部分(瓶口、螺纹)已经成型;第二步:人工将管坯放在蜂窝状加热器或自动循环加热传送带上加热调温,然后移到吹瓶机用压缩空气吹制成型。 优点:设备较简单,投资较少。瓶口较平整,密封良好。品种开发快,模具费用较低,成本价格中、低。 缺点:注射管坯与吹塑成型分步进行,易传递污染,菌检难保证,产品同一性差,不太适应大批量生产。 (3)一步法“注-吹”one step injection-blow moulding。“注射、吹塑”在同一台机器上完成。根据不同机种又分为三工位和二工位“注-吹”。三工位“注-吹”制瓶机三个工位以120°角成等边三角形分布,第一工位为注射成型工位,第二工位为吹塑成型工位,第三工位为脱瓶工位。三个工位可同时运行,生产效率高,周期短,而且可与传送带连接自动计数包装,真正实现药用塑料瓶生产全过程中与人手“无接触”,确保产品卫生洁净。二工位“注-吹”塑料机二工位可上、下或前后排列;第一工位为注射成型工位,第二工位为吹塑成
药用辅料甜菊素质量标准的研究
药用辅料甜菊素质量标准的研究 摘要:目的:根据《药用辅料管理办法》的有关要求,对作为药用辅料的甜菊 素进行质量研究,为其质量标准的提高提供参考,对进一步规范厂家药用辅料甜 菊素的生产、认证、检查提出意见及建议。方法:参考《中国药典》(2015 版) 及《药用辅料管理办法》的相关要求,从结构确证、稳定性实验、性状、鉴别、 检查及含量测定等方面进行研究。结果:甜菊素的光谱图、色谱图均与对照品一致,长期加速实验结果表明甜菊素质量稳定,甜菊素各项质量指标符合《中国药典》(2015 版)药用辅料甜菊素的质量标准要求。结论:研究的甜菊素结构明确,质量稳定且符合标准要求,可用《中国药典》(2015 版)药用辅料甜菊素标准进 行质量控制。 关键词:药用辅料;甜菊素;质量标准 The pharmaceutical excipients stevia qualities criteria Xu Jianghong,Zhang Ruoliang,Liu Chunmei,Huang Weibo,Yu Jing (Ganzhou Municipal Center for Food and Drug Contro 341000) ABSTRACT:OBJECTIVE:According to the relevant requirements of the measures for the administration of the pharmaceutical excipients,the value of the stevia as pharmaceutical excipients quality were studied in order to provide a reference for the improvement of its quality standard and to further standardize the manufacturer pharmaceutical excipients stevia grain production,certification,inspection opinions andsuggestions are put forward. METHODS:According to the Chinese pharmacopoeia (2015 edition)and "medicinal supplementary material management method",the requirements of the relevant confirmed from the structure,stability testing,character,identification,examination and content determination were studied. RESULTS:The spectrum and chromatogram of steviosin of are consistent with the reference substance,accelerate the experimental results show that steviosin qualities for a long time stability,steviosin,the quality indicators in line with the "Chinese pharmacopoeia"(2015 edition)pharmaceutical excipients steviosin grain quality standard requirements. CONCLUSION:The structure of steviosin is clear,stable quality and conform to the requirements of the standards,the available "China pharmacopoeia"(2015 edition)pharmaceutical excipients stevia standards for quality control. KEY WORDS:Pharmaceutical excipient;Steviosin;Qualities criteria 甜菊素是从天然甜料植物甜叶菊的甜叶中提取的一类高甜度、低热值、非发 酵性、对人体无副作用的天然甜味剂,对肥胖病、糖尿病、高血压病、心脏病、 龋齿等也有一定的辅助治疗作用。甜菊素的甜度是蔗糖的300倍,而其热值却为 蔗糖的1/300,所以目前不少药品生产厂家把甜菊素作为一种甜味剂替代蔗糖。 甜菊素是经我国卫生部、食品药品监督管理局批准使用的最接近蔗糖口味的天然 低热值矫味剂和甜味剂。它是继甘蔗甜菜糖之外第3种有开发价值和健康推崇的 天然蔗糖替代品,被国际上誉为“世界第三蔗糖”。随着人们对健康的关注程度越 来越重,甜菊素作为一种新型的药用辅料必将具有良好的市场前景。甜菊素收载 于《中国药典》2015年版,属已有国家标准的药用辅料,本文分别采用紫外吸收 光谱法、红外光谱法、高效液相色谱法对甜菊素的化学结构及组份进行了确证,
中药材质量标准海龙
中药材质量标准海龙 The latest revision on November 22, 2020
中药材质量标准 中文名海龙 汉语拼音Hailong 英文名SYNGNATHUS 来源本品为海龙科动物刁海龙Solenognathus hardwickii(Gray)、多棘刁海龙Syngnathus guntheri Dunker的干燥体。 性状刁海龙体狭长侧扁,全长30~50cm。表面黄白色或灰褐色。头部具 管状长吻,口小,无牙,两眼圆而深陷,头部与体轴略呈钝 角。躯干部宽3cm,五棱形,尾部前方六棱形,后方渐细,四 棱形,尾端卷曲。背棱两测各有l列灰黑色斑点状色带。全 体被以具花纹的骨环及细横纹,各骨环内有突起粒状棘。胸 鳍短宽,背鳍较长,有的不明显,无尾鳍。骨质,坚硬。气 微腥,味微咸。 多棘刁海龙体长条形而侧扁,中部略粗壮。长36~47cm,中部直径 3~5cm。头部具管状长咀,两眼内陷,头鳃部密被棘状突 起。表面黄白色或灰棕色,全体有类圆形突起的“雪花样” 纹理与横纹组成的图案状花纹。躯干部具7条纵棱,其中两 侧棱隆起不明显,有骨环 25~26个。尾部前段具6条纵棱,后段类方形,具4条纵棱,尾端卷 曲,有骨环66~68个,无尾鳍。骨质,坚硬。气微腥,味微 咸。 鉴别 检查酸不溶性灰分不得过%(中国药典2000年版一部附录IX K)。 对二氯苯照气相色谱法(中国药典2000年版一部附录ⅥE)测定,应不得检出。 色谱条件与系统适用性试验以硅酮(OV-17)为固定相,涂布浓度为%。柱温85℃。理论板数按对二氯苯计算,应不低于2000。 对照品溶液的制备:取对二氯苯对照品约20mg,精密称定,置100ml量瓶中,加环己烷溶解并稀释至刻度,摇匀。 供试品溶液的制备:将本品粉碎成粗粉,混匀,取样品5g,精密称定,精密加入环己烷10ml,密塞,超声处理15分钟,放冷后滤 过,即得。 测定法精密量取对照品溶液及供试品溶液各1μl,分别注入气相色谱仪,测定,计算,即得。 浸出物 含量测定 炮制除去灰屑。用时捣碎或切段。 性味与归经甘,温。归肝、肾经。 功能与主治温肾壮阳,散结消肿。用于阳痿遗精,症瘕积聚,瘰疬痰核,跌扑损伤;外治痈肿疔疮。
药用辅料申报资料要求
附件2 药用辅料申报资料要求(试行) 品种名称:XXXXX 申请人:XXXXX 应用情况:□境内外上市制剂中未使用过的药用辅料 □境外上市制剂中已使用而在境内上市制剂中未使用过的 药用辅料 □境内上市制剂中已使用,未获得批准证明文件或核准编 号的药用辅料 □已获得批准证明文件或核准编号的药用辅料改变给药途 径或提高使用限量 □国家食品药品监督管理总局规定的其他药用辅料拟用制剂给药途径:□注射□吸入□眼用□局部及舌下□透皮□口服□其他 来源:□动物或人□矿物□植物□化学合成□其他 注册申请人名称:盖章 法定代表人:签名
一、申报资料项目 1 企业基本信息 1.1 企业名称、注册地址、生产地址1.2 企业证明性文件 1.3 研究资料保存地址 2 辅料基本信息 2.1 名称 2.2 结构与组成 2.3 理化性质及基本特性 2.4 境内外批准信息及用途 2.5 国内外药典收载情况 3 生产信息 3.1 生产工艺和过程控制 3.2 物料控制 3.3 关键步骤和中间体的控制 3.4 工艺验证和评价 3.5 生产工艺的开发 4 特性鉴定 4.1 结构和理化性质研究 4.2 杂质研究 4.3 功能特性 5 质量控制
5.1 质量标准 5.2 分析方法的验证 5.3 质量标准制定依据 6 批检验报告 7 稳定性研究 7.1 稳定性总结 7.2 稳定性数据 7.3 辅料的包装 8 药理毒理研究 二、申报资料正文及撰写要求 1 企业基本信息 1.1 企业名称、注册地址、生产地址 提供企业的名称、注册地址、生产厂、生产地址。 生产地址应精确至生产车间、生产线。 1.2 企业证明性文件 境内药用辅料生产企业需提交以下证明文件: (1)企业营业执照复印件。如企业同时持有其他类型许可证,也应同时提供其复印件,如《药品生产许可证》《食品添加剂生产许可证》及相关认证文件等。 (2)对于申请药用明胶空心胶囊、胶囊用明胶和药用明胶的国内生产企业,需另提供:①申请药用空心胶囊的,应提供明胶的合法来源证明文件,包括药用明胶的批准证明文件、标准、
药用包装材料质量标准ISO15378(中文)
ISO 15378: 2006 标准的中文版内容 国际标准化组织 /第76技术委员会 (IS0/TC76) 于2003年制定了 1S0 15378国际标准草案 (Draft of interrfational standard ,D1S),标题是:《药品初包装材料 ISO 9001:2008 应用的专用要求,包含生产质量管理规范(GMP) 》。2006 年,形成了借鉴 ISO 9001:2008 质量体系的《药用包装材料质量标准 ISO 15378 : 2006 》初稿。这个国际标准的制定说明了国际社会对药包材生产企业实施质量管理的重视。此处择其要简介其主要内容 (略去的有些内容可参见 ISO 9001: 2008 标准) ( 一 ) 引言 引言部分包括 :总则;过程方法;与 ISO 9004 的关系;与其他管理体系的相容性。 0.1 总则 本标准把 GMP原理和 QMS质量管理体系规定的要求应用于药品的初包装材料。由于初包装材料与药品直接接触,组织 (企业) 对初包装材料的生产和质量控制中的领会 GMI'原理对于患者使用药品时的安全性是非常重要的。药用包装材料应用 GMP应能确保这些材料满足制药工业的需求。 采用 QMS应当是组织的一项战略性决策。一个组织 QMS的设计和实施受各种需求、具体目标、所提供的产品、所采用的过程以及该组织的规模和结构的影响。 . ISO 15378 的主要目的是规定协商的初包装材料的要求。它包括一些初包装材料的专用要求,这些要求出自药品生产、控制等生产质量管理规范。 0.2 过程方法 本标准鼓励在建立、实施 QMS以及改进其有效性时采用过程方法,通过满足顾客要求,增强顾客满意。 过程方法在 QMS中应用时,强调以下方面的重要性: (1)理解并满足要求 ;
药材原料质量标准
原料肉苁蓉的质量标准 【来源】本品为列当科植物肉苁蓉或管花肉苁蓉()的干燥带鳞叶的肉质茎。春季苗刚出去时或秋季冻土之前采挖,除去茎尖。切段,晒 干。 【性状】本品呈扁圆柱形,稍弯曲,长~,直径~。表面棕褐色或灰棕色,中间有淡棕色小点(维管束)排列成波状环纹;周边呈灰黑色鳞 片状;体重,质硬,微有柔性,不易折断,断面棕褐色,气微, 味甜、微苦。 【鉴别】取本品粉末,加甲醇,超声处理分钟,滤过,滤液浓缩至近干,残渣加甲醇使溶解作为供试品溶液。另取松果菊苷对照品、毛蕊 花糖苷对照品。分别加甲醇制成每含的溶液,作为对照品溶液。 照薄层色谱法(附录)试验,吸取上述三种溶液各μ,分别点于 同一聚酰胺薄层板上,以甲醇醋酸水()为展开剂,展开,取出, 晾干,置紫外光灯()下检视。供试品色谱中,在与对照品色谱 相应的位置上,显相同颜色的荧光斑点。 【检查】水分照水分测定法(附录)第一法测定,不得过。 总灰分不得过(附录)。 酸不溶性灰分不得过(附录)。 【含量测定】照醇溶性浸出物测定法项下的冷浸法(附录)测定,用乙醇作溶剂,不得少于。 肉苁蓉照高效液相色谱法(附录)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;乙腈甲醇醋酸溶液(::)为流动相;检测波长为。理论板数按松果菊苷峰计算应不低于。 对照品溶液的制备精密称取松果菊苷对照品和毛蕊花糖苷对照品适量,分别加流动相制成每含松果菊苷和毛蕊花糖苷的溶液,即得. 供试品溶液的制备取本品粉末(过四号筛)约,精密称定,置棕色量瓶中,精密加流动相,称定重量,浸泡小时,超声处理(功率,频率)分钟,放冷,再称定重量,用流动相补足减失的重量,摇匀,离心,取上清液置棕色量瓶中,即得。 测定法分别精密吸取对照品溶液~与供试品溶液~,注入液相色谱仪,测定,即得。 本品按干燥品计算,含松果菊苷()和毛蕊花糖苷()的总量不得少于。 管花肉苁蓉照高效液相色谱法(附录)测定。 色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;甲醇甲酸溶液(:)为流动相;检测波长为。理论板数按松果菊苷峰计算应不低于。 对照品溶液的制备精密称取松果菊苷对照品适量,置棕色量瓶中,用甲醇制成每含的溶液,即得。 供试品溶液的制备取本品粉末(过四号筛)约,精密称定,置棕色量瓶中,精密加甲醇,密塞,称定重量,浸泡小时,超声处理(功率,频率)分钟(o以下),取出,放冷,再称定重量,用甲醇补足减失的
药用塑料瓶的质量监控与检测方法
药用塑料瓶的质量监控与检测方法 摘要: 药用塑料瓶以其质量轻、强度高、密封性和耐压性好等优点,正逐步替代药用玻璃瓶,被广泛应用于口服固体药和液体药的包装领域。但由于药品成分复杂、性能各异,储藏条件苛刻,所以对塑料瓶的要求也更加严格。相应标准的出台,意味着企业更需要严格执行对药用塑料瓶的质量监控与检测工作。本文将依据标准要求,简单介绍一下药用塑料瓶的阻隔性能与密封性测试方法。 关键词:药用塑料瓶、密封性、扭力、水蒸气渗透 作者:济南兰光机电技术有限公司 背景: 药品的质量安全直接影响国民健康,包装作为药品的重要组成部分,在生产出厂后的质量保护方面扮演着重要角色。药用塑料瓶以其质量轻、强度高、密封性和耐压性好等优点,正逐步替代药用玻璃瓶,被广泛应用于口服固体药和液体药的包装领域。但由于药品成分复杂、性能各异,储藏条件苛刻,所以对塑料瓶的要求也更加严格。 生产的药用塑料瓶一定要经过质量部门的相关检测,以保证生产的药用塑料瓶达到国家的出厂规范,在医药的运用过程中确保不会出现质量事故,确保药品安全。2002年,我国国家药品监督管理局发布了口服液体药用塑料瓶的国家药品包装容器标准,分别为YBB00082002《口服液体药用聚丙烯瓶-试行版》、YBB00092002《口服液体药用高密度聚乙烯瓶-试行版》、YBB00112002《口服固体药用聚丙烯瓶》等。这些标准的出台,意味着企业需要严格执行对药用塑料瓶的质量监控与检测工作。 药用塑料瓶目前主要采用PP、HDPE、PET三种材料,本文将从上述材料药用塑料瓶的标准要求出发,简单介绍一下药用塑料瓶的阻隔性能与密封性测试方法。 阻隔性能检测 阻隔性能是指包装材料对气体、液体等渗透物的阻隔作用。测试项目包括气体与水蒸气透过性能检测两类。氧气与水蒸气是影响药品质量的重要因素,若药用塑料瓶的阻隔性能不好,必然会导致药品因接触过多的氧气与水蒸气产生变质问题,这就要求企业对药用塑料瓶的阻隔性能进行检测。 (1)氧气透过率检测
注射剂产品直接接触药品的包装材料和容器的选择考虑 20081031
注射剂产品直接接触药品的包装材料和容器的选择考虑 发布日期20081031 栏目化药药物评价>>化药质量控制 作者审评三部霍秀敏 直接接触药品的包装材料和容器是药品不可分割的一部分,它伴随药品生产、流通及使用的全过程。由于包装材料、容器的组成、药品所选择的原辅料及生产工艺的不同,药品包装材料和容器中有的组份可能会被所接触的药品溶出、或与药品发生互相作用、或被药品长期浸泡腐蚀脱片而直接影响药品的质量;而且,有些对药品质量及人体的影响具有隐患性(即通过对药品质量及人体的常规检验不能及时发现的问题)。例如,安瓿、输液瓶(袋),如果不是针对不同药品采用不同的处方和生产工艺进行选择,常常会有药品包装材料和容器中的组份被溶出及玻璃脱片现象,这些影响在一般的常规药检时不能被发现;再例如,天然橡胶塞中溶出的异性蛋白对人体可能是致热源,溶出的吡啶类化合物是致癌、致畸、致突变的肯定因素,而细微的玻璃脱片是堵塞血管形成血栓或肺肉芽肿隐患等等。从另一个方面讲,由于药品的种类多且有效活性基团复杂,不同药品与直接接触药品的包装材料和容器之间的相互影响也不同,所以,一种包装材料和容器适用于所有的药品,或者一种药品可以采用任何可获得的包装材料和容器都是存在巨大的质量和安全性隐患的。药品是一种特殊的商品,特别是注射剂产品,其质量和由包装材料和容器引起的安全性隐患要高于口服剂型,所以对注射剂产品的直接接触药品的包装材料和容器的选择,不仅要考虑包装材料和容器是否能满足药品本身应能达到的无菌保证水平的要求,同时更要关注直接接触药品的包装材料和容器与药品之间的相互作用。 1 我国药包材生产企业的现状与管理要求 我国药包材生产企业和药包材产品相对落后。虽然,现有药包材生产企业约1000家,生产
中药制定标准
一、中药材质量标准 (一)质量标准 包括名称、汉语拼音、药材拉丁名、来源、性状、鉴别、检查、浸出物、含量测定、炮制、性味与归经、功能与主治、用法与用量、注意及贮藏等项。有关项目内容的技术要求如下: 1.名称、汉语拼音、药材拉丁名按中药命名原则要求制定。 2.来源 来源包括原植(动、矿)物的科名、中文名、拉丁学名、药用部位、采收季节和产地加工等,矿物药包括矿物的类、族、矿石名或岩石名、主要成分及产地加工。上述的中药材(植、动、矿等)均应固定其产地。 (1)原植(动、矿)物需经有关单位鉴定,确定原植(动)物的科名、中文名及拉丁学名;矿物的中文名及拉丁名。 (2)药用部位是指植(动、矿)物经产地加工后可药用的某一部分或全部。 (3)采收季节和产地加工系指能保证药材质量的最佳采收季节和产地加工方法。 3.性状 系指药材的外形、颜色、表面特征、质地、断面及气味等的描述,除必须鲜用的按鲜品描述外,一般以完整的干药材为主;易破碎的药材还须描述破碎部分。描述要抓住主要特征,文字要简练,术语需规范,描述应确切。 4.鉴别 选用方法要求专属、灵敏。包括经验鉴别、显微鉴别(组织切片、粉末或表面制片、显微化学)、一般理化鉴别、色谱或光谱鉴别及其它方法的鉴别。色谱鉴别应设对照品或对照药材。 5.检查 包括杂质、水分、灰分、酸不溶性灰分、重金属、砷盐、农药残留量、有关的毒性成分及其它必要的检查项目。 6.浸出物测定 可参照《中国药典》附录浸出物测定要求,结合用药习惯、药材质地及已知的化学成分类别等选定适宜的溶剂,测定其浸出物量以控制质量。浸出物量的限(幅)度指标应根据实测数据制订,并以药材的干品计算。 7.含量测定 应建立有效成分含量测定项目,操作步骤叙述应准确,术语和计量单位应规范。含量限(幅)度指标应根据实测数据制订。 在建立化学成分的含量测定有困难时,可建立相应的图谱测定或生物测定等其它方法。
国家食品药品监督管理局关于印发加强药用辅料监督管理有关规定的.
国家食品药品监督管理局关于印发加强药用辅料监督管理有关规定的通知 国食药监办 [2012]212号 2012年 08月 01日发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局 : 为进一步加强药用辅料生产和使用管理, 保证药品质量, 依据《中华人民共和国药品管理法》及其实施条例等法律法规, 国家食品药品监督管理局组织制定了《加强药用辅料监督管理的有关规定》。现印发你们,请督促行政区域内相关企业遵照执行,并做好监督检查工作。 国家食品药品监督管理局 2012年 8月 1日 加强药用辅料监督管理的有关规定 药用辅料是药品的重要组成部分,直接影响药品的质量。为进一步加强药用辅料生产、使用的监管,确保药品质量安全,依据《中华人民共和国药品管理法》及其实施条例、《国务院关于加强食品等产品安全监督管理的特别规定》、《药品生产监督管理办法》、《药品注册管理办法》、《药品生产质量管理规范》等相关法律法规规章,特规定如下: 一、药品制剂生产企业必须保证购入药用辅料的质量 (一药品制剂生产企业是药品质量责任人。必须切实加强药品生产质量管理,确保药品质量安全。必须严格药用辅料使用的管理, 按照药品监督管理部门核准的处方工艺, 使用符合要求的药用辅料生产药品。凡因违法违规使用药用辅料引发的药品质量问题, 药品制剂生产企业必须承担主要责任。 (二药品制剂生产企业必须健全质量管理体系。应确保质量管理部门有效履行质量保证和质量控制职责, 企业负责人及其他部门人员不得干扰或妨碍质量管理部门履行职责。确定药用辅料供应商应进行审计并经企业质量管理部门批准。
(三药品制剂生产企业应加强药用辅料供应商审计。应按照《药品生产质量管理规范 (2010年修订》有关要求,对药用辅料生产企业定期进行质量评估,对药用辅料生产企业的质量体系进行质量审计和回顾分析,并建立所有购入药用辅料及供应商的质量档案。 (四药品制剂生产企业必须对所使用的药用辅料质量严格把关。凡购入的药用辅料, 都必须按照药品批准注册时核准的质量标准进行检验, 确保符合药用要求。对已颁布国家药品标准的药用辅料,必须符合国家药品标准的要求。 (五药品制剂生产企业应与主要药用辅料供应商签订质量协议。随时掌握所使用药用辅料的变更情况,研究和评估变更对药品质量的影响,并按照《药品注册管理办法》的要求进行申报。 二、药用辅料生产企业必须保证产品的质量 (六药用辅料生产企业必须对产品质量负责。应严格执行《药用辅料生产质量管理规 范》 , 健全企业质量管理体系, 加强对生产所用原材料的供应商审计, 严格原材料质量控制, 按照产品注册核准的处方工艺组织生产, 规范产品批号的编制, 保证产品质量稳定。对未取得批准文号且历史沿用的药用辅料, 应按照与药品制剂生产企业合同约定的质量协议组织生产。 (七药用辅料生产企业必须保证产品质量。应按注册批准的或与药品制剂生产企业合同约定的质量标准, 对每批产品进行全项检验,合格后方可入库、销售。对已颁布国家药品标准的药用辅料,必须符合国家药品标准的有关要求。产品放行前,所有生产文件和记录, 包括检验数据均应经质量管理部门审查并符合要求,不符合要求不得放行出厂。 (八药用辅料生产企业应配合药品制剂生产企业开展供应商审计。若发生生产工艺、原材料来源等可能影响药用辅料质量的变更时, 应主动开展相应的评估, 及时通报药品制剂生产企业。 三、药品监督管理部门对药用辅料实施分类管理
- 药用包装材料质量标准ISO15378(中文)
- 药品包材质量标准
- GMP包装材料质量标准
- 药品包装用材料
- 药品包装材料与药相容性试验指导原则
- 国家药品包装容器材料标准
- 药用包装材料质量标准
- 药用包装材料质量标准ISO15378(中文)
- 包装材料质量管理规程
- 药品与包装材料的相容性研究
- 药用包装材料质量标准
- GMP包装材料质量标准(1)
- 药用包装材料质量标准ISO中文
- 药用包装材料高质量实用标准ISO15378(中文)
- 药品泡罩包装材料质量标准一致性研究
- 各类医用包装材料质量技术参数表
- 药用包装材料质量标准
- 注射剂产品直接接触药品的包装材料和容器的选择考虑 20081031
- GMP包装材料质量标准
- 包装印刷包装材料质量标准、检验操作规程