Digitalmicrograph也可以做颗粒尺寸分析
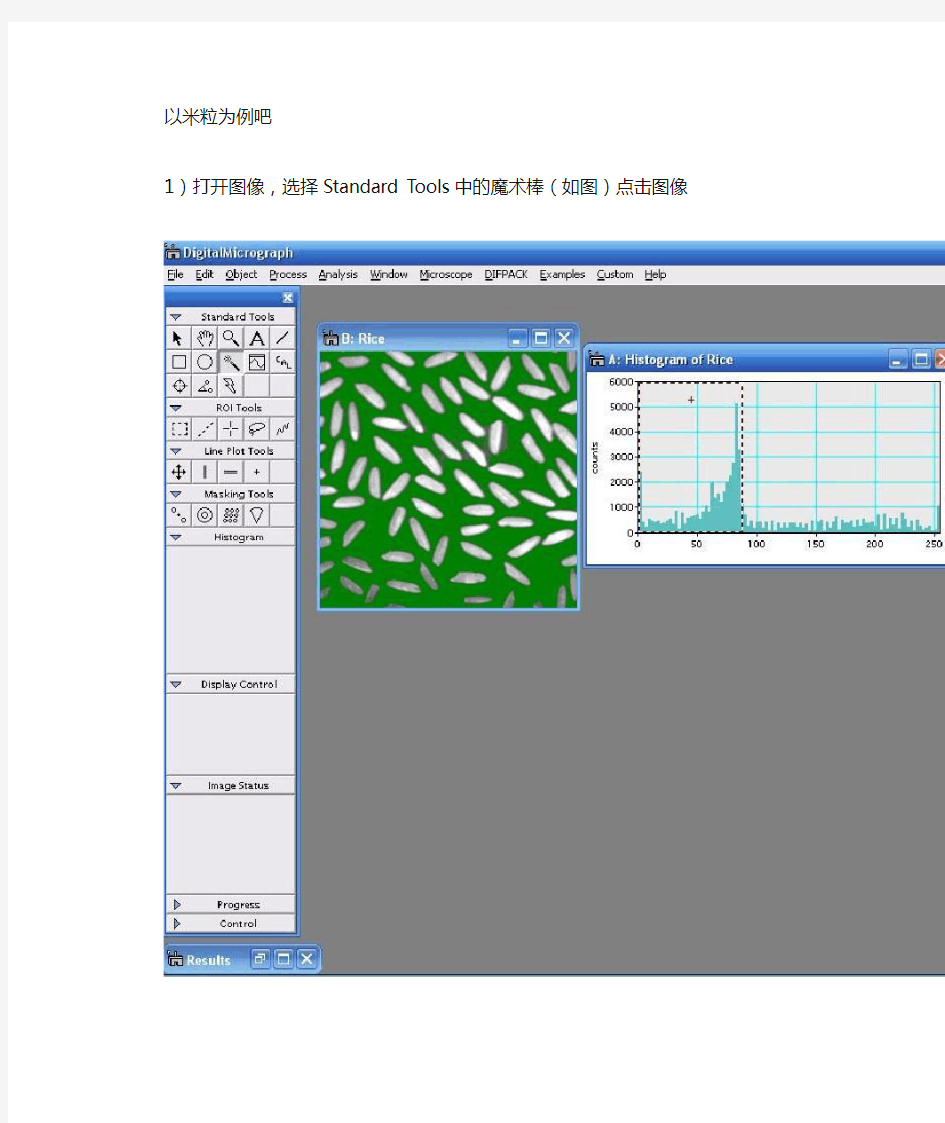
以米粒为例吧
1)打开图像,选择Standard Tools中的魔术棒(如图)点击图像
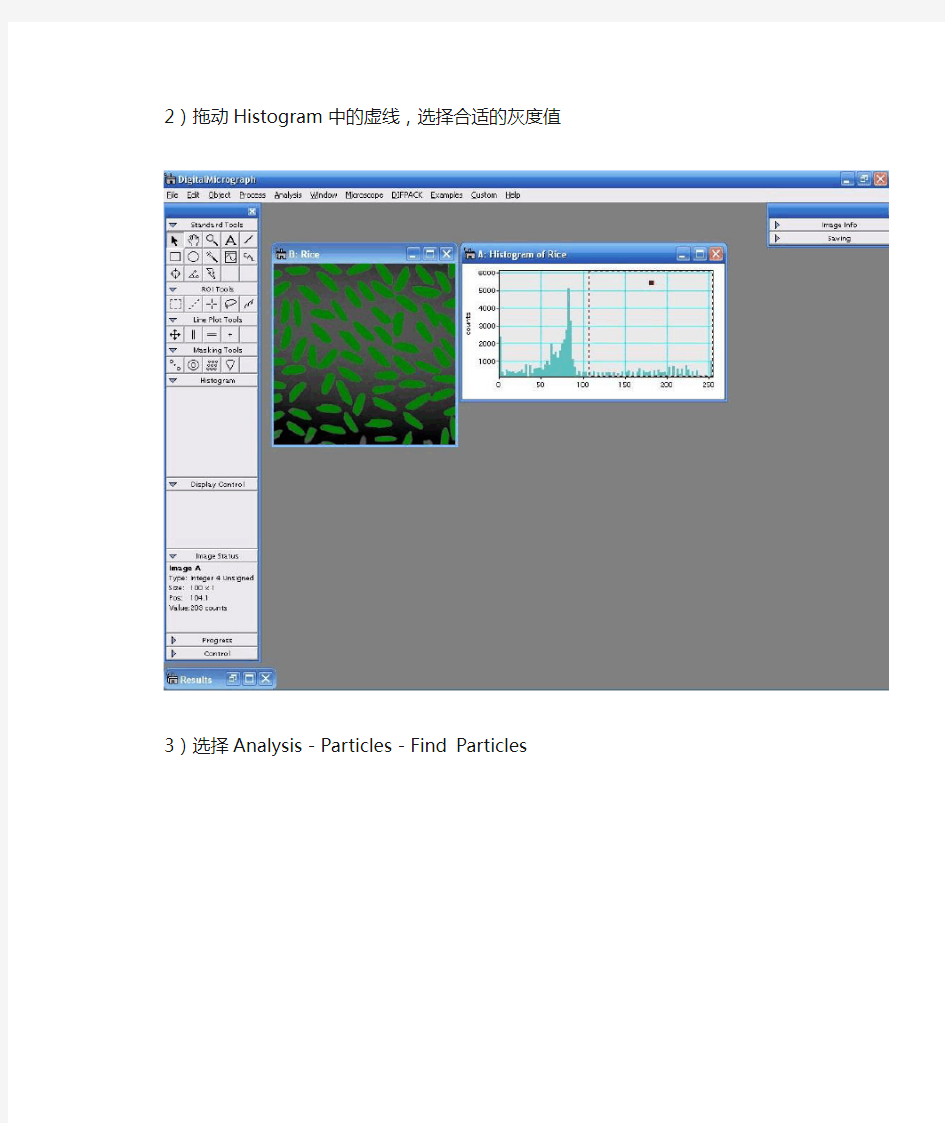
2)拖动Histogram中的虚线,选择合适的灰度值
3)选择Analysis-Particles-Find Particles
4)选择Analysis-Particles-Analyze Particles
搞定!
颗粒大小分析
附录 土 工 试 验 实验四 颗粒大小分析试验 (一)概述 试验目的是使用比重计法测定土的各种粒组占该土总质量的百分数,并据此绘制颗粒大小分配曲线。比重计法适用于分析粒径小于0.075mm 的土样,若试样中还有大于0.075mm 的粒径时,应联合使用比重计法和筛析法。 (二)试验原理 比重计法是将一定质量的试样加入4%浓度的六偏磷酸钠10mL 混合成1000mL 悬液,并使悬液中的土粒均匀分布。此时悬液中不同大小的土粒下沉速度快慢不一。一方面可由斯笃克(Stokes ,1845)定律计算悬液中不同大小土粒的直径,另一方面用比重计测定其相应不同大小土粒质量的百分数。 1.斯笃克定律 根据斯笃克定律各种土粒在悬液中的下沉速度与其直径大小、比重和液体的动力粘滞系数有关。在时间t 内的下沉速度v 为: 2 4101800)(gd d d t L v w wt s η ρ?-== 或 t L k d = g d d k w wt s ρη)(1018004-?= 式中 v ——土颗粒下沉速度,cm/s ; η——纯水的动力粘滞系数,10-6kPa·s d ——土颗粒粒径,mm ; g ——重力加速度,981cm/s 2; d s ——土粒的比重; ρw ——4℃时水的密度,g/cm 3; d wt ——温度T ℃时水的比重; L ——某一时间t 内土粒的沉降距离,cm ; t ——土粒沉降的时间,s ; k ——粒径计算系数。 为了简化计算,用图附4.1的斯笃克列线图,便可求得粒径d 值。此时,悬液中在L 范围内所有土粒的直径都比算得的d 值小,而大于d 的土粒都下沉到比L 大的深度处。 2.悬液中土粒质量的百分数 附4.1
XRD晶粒尺寸计算
XRD晶粒尺寸分析 很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A。这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,如果这个方向上有M个单胞,而且这个方向上的晶面间距为d,则测得的尺寸就是Md。如果某个方向(HKL)的单胞数为N,晶面间距为d1,那么这个方向的尺寸就是Nd1。由此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B 如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C 为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来也特别麻烦(现在好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽计算出来的“粒径”总是那么小,何乐而不为呢?我私下地觉得吧,这些人在偷换概念。久而久之,大家也就接受了。 为了这个事吧,有些人就问了,既然做出来的纳米材料的“粒径”是这么小,那么有没有办法在做SEM或TEM时将团聚在一起的小晶粒分开呢?确实分不开,分得开的是一个个的晶粒,分不开的是亚晶。 D 至于为什么通过衍射峰宽测出来的“粒径”为什么总是那么小,还有一个原因。实际上吧,使衍射峰变宽的原因可能有两个,一是晶粒变小了,另一个原因是晶粒内部存在“微观应变”。打个比方吧,甲乙两个人同时做一件事,结果把功劳算到甲一个人头上,当然这个人的功劳就大了(功能劳大就峰宽,峰越宽晶粒就越细)。有时候发现,有个别人在有意无意地避口不谈乙的功劳。 E 为什么允许将亚晶尺寸称为“粒径”呢?称为径,必假定晶粒为“球形”,从而假定了不论从哪个晶面去测都会是相同的,即忽略了A 所说的那种差别。事实上,这种不同方向的尺寸差异在很多情况下确实可以忽略。但是,也有一些特殊情况是不可以的。下面我们再谈。 注意这两个假定,这就是为什么很多人都说,XRD测出来的粒径不可靠,总是小于SEM和TEM量出来的值。因为概念都不相同,它们怎么可能相同呢? 既然大家都说是粒径,那么要怎么样来算粒径呢? 我们先来看一个简单的问题。 怎么做拟合?
如何进行晶粒度分析
教你如何进行晶粒度分析 金属晶粒的尺寸(或晶粒度)对其在室温及高温下的机械性质有决定性的影响,晶粒尺寸的细化也被作为钢的热处理中最重要的强化途径之一。因此,在金属性能分析中,晶粒尺寸的估算显得十分重要。那么根据一张金相照片我们能从中得到哪些信息呢? 首先来看看这一段小视频 视频:晶粒度分析 一、晶粒度概述 晶粒度表示晶粒大小的尺度。金属的晶粒大小对金属的许多性能有很大影响。晶粒度的影响,实质是晶界面积大小的影响。晶粒越细小则晶界面积越大,对性能的影响也越大。对于金属的常温力学性能来说,一般是晶粒越细小,则强度和硬度越高,同时塑性和韧性也越好。 二、测定平均晶粒度的基本方法 一般情况下测定平均晶粒度有三种基本方法:比较法、面积法、截点法。具体如下 1、比较法:比较法不需计算晶粒、截矩。与标准系列评级图进行比较,用比较法评估晶粒度时一般存在一定的偏差(±0.5级)。评估值的重现性与再现性通常为±1级。 2、面积法:面积法是计算已知面积内晶粒个数,利用单位面积晶粒数来确定晶粒度级别数。该方法的精确度中所计算晶粒度的函数,通过合理计数可实现±0.25级的精确度。面积法的测定结果是无偏差的,重现性小于±0. 5级。面积法的晶粒度关键在于晶粒界面明显划分晶粒的计数
图:面积法 3、截点法:截点数是计算已知长度的试验线段(或网格)与晶粒界面相交截部分的截点数,利用单位长度截点数来确定晶粒度级别数。截点法的精确度是计算的截点数或截距的函数,通过有效的统计结果可达到±0.25级的精确度。截点法的测量结果是无偏差的,重现性和再现性小于±0.5级。对同一精度水平,截点法由于不需要精确标计截点或截距数,因而较面积法测量快。 同心圆测量线(截点法) 三、金相图具体案例分析 以上只是大致的测定方法太过笼统,如果真的拿到一个具体的微观照片,我们该怎么做呢?下面我们来看一下具体操作与计算方法。
xRD晶粒尺寸分析
xRD晶粒尺寸分析
XRD晶粒尺寸分析 注:晶粒尺寸和晶面间距不同 计算晶粒大小:谢乐公式:D=kλ/βcosθ D—垂直于反射晶面(hkl)的晶粒平均粒度D是晶粒大小 β--(弧度)为该晶面衍射峰值半高宽的宽化程度 K—谢乐常数,取决于结晶形状,常取0.89 θ--衍射角 λ---入射X射线波长(?) 计算晶面间距:布拉格方程:2dsinθ=nλd是晶面间距。 此文档是用XRD软件来分析晶粒尺寸,用拟合的办法,而不是用谢乐公式 很多人都想算算粒径有多大。 其实,我们专业的术语不叫粒径,而叫“亚晶尺寸”,它表征的并不是一个颗粒的直径。 A 这么说吧,粉末由很多“颗粒”组成,每个颗粒由很多个“晶粒”聚集而成,一个晶粒由很多个“单胞”拼接组成。X射线测得的晶块尺寸是指衍射面指数方向上的尺寸,
如果这个方向上有M 个单胞,而且这个方向上的晶面间距为d ,则测得的尺寸就是Md 。如果某个方向(HKL )的单胞数为N ,晶面间距为d 1,那么这个方向的尺寸就是Nd 1。由 此可见,通过不同的衍射面测得的晶块尺寸是不一定相同的。 B 如果这个晶粒是一个完整的,没有缺陷的晶粒,可以将其视为一个测试单位,但是,如果这个晶粒有缺陷,那它就不是一个测试单位了,由缺陷分开的各个单位称为“亚晶”。比如说吧,如果一个晶粒由两个通过亚晶界的小晶粒组成(称为亚晶),那么,测得的就不是这个晶粒的尺寸而是亚晶的尺寸了。 C 为什么那么多人喜欢抛开专业的解释而用“粒径”这个词呢?都是“纳米材料”惹的祸。纳米晶粒本来就很小,一般可以认为一个纳米晶粒中不再存在亚晶,而是一个完整的晶粒,因此,亚晶尺寸这个术语就被套用到纳米晶粒的“粒径”上来了。实际上,国家对于纳米材料的粒径及粒径分布的表征是有标准的,需要用“小角散射”方法来测量。比如,北京钢铁研究总院做这个就做了很长时间。但是呢,一则,做小角散射的地方还不多,做起来也特别麻烦(现在好一些了,特别是对光能自动一些了),所以,很少有人去做,而且,用衍射峰宽计算出来的“粒径”总
实验一、颗粒大小分析试验(比重计法)
实验一、颗粒大小分析试验(比重计法) 颗粒大小分析试验是测定干土中各种粒组所占该土总质量的百分数,借以明确颗粒大小分布情况,供土的分类与概略判断土的工程性质及选料之用。根据土的颗粒大小及级配情况常用的方法有筛分法与比重计法,筛分法适用于分析粒径大于0.074mm 的土;比重计法适用于粒径小于0.074mm的土。当土中兼有上述两类粒径时,则应联合使用筛析法与比重计法。 一、基本原理 密度计法是静水沉降分析法的一种,只适用于粒径小于0.075mm的土样。密度计法是将一定量的土样(粒径<0.075mm)放在量筒中,然后加纯水,经过搅拌,使土的大小颗粒在水中均匀分布,制成一定量的均匀浓度的土悬液(1000mL)。静止悬液,让土粒沉降,在土粒下沉过程中,用密度计测出在悬液中对应于不同时间的不同悬液密度,根据密度计读数和土粒的下沉时间,就可计算出粒径小于某一粒径d(mm)的颗粒占土样的百分数。 二、仪器设备 1、密度计 目前通常采用的密度计有甲、乙两种,这两种密度计的制造原理及使用方法基本相同,但密度计的读数所表示的含义则是不同的,甲种密度计读数所表示的是一定量悬液中的干土质量;乙种密度计读数所表示的是悬液比重。 (1)甲种密度计,刻度单位以在20oC时每1000mL悬液内所含土质量的克数来表示,刻度为-5~50,最小分度值为0.5。 (2)乙种密度计,刻度单位以在20oC时悬液的比重来表示,刻度为0.995~1.020,最小分度值为0.0002。 2、量筒2个:容积1000mL; 3、三角烧瓶:容积500ml 4、煮沸设备:电热器、锥形烧瓶; 5、分散剂:4%六偏磷酸钠或25%氨水; 6、其他:搅拌棒、温度计、研钵、秒表、烧杯、瓷皿、天平等。 三、操作步骤 1、密度计的校正 密度计在制造过程中, 其浮泡体积及刻度往往不易准确, 况且, 密度计的刻度是 以20 C的纯水为标准的。由于受实验室多种因素的影响,密度计在使用前应对刻度、弯液面、土粒沉降距离、温度、分散剂等的影响进行校正。 (1)土粒沉降距离校正
晶粒度研究分析
晶粒度分析
————————————————————————————————作者:————————————————————————————————日期:
DEFROM-3D之晶粒度分析模拟1.创建一个新问题 在主窗口中选中一个DB文件,单机后处理的Microsoftstructure按钮,打开 DEFORM-MICROSTRUCTURE窗口,单击Add project按钮增加计划。 2. 追踪选项设置 点击define按钮,在坯料上选取5个点,如图2所示
单机next按钮,在追踪界面选中No单选按钮,点击next。 3. 离散点阵设置 在离散点阵界面,类型选中Celluar Automata单选按钮(即CA模型),几何选中Square单选按钮,行和列分别设置为50,绝对尺寸为1,如图3所示 4. 边界条件设置 在边界条件界面,保持默认设置即可 5. 晶粒边界条件设置 在晶粒边界选项界面,设定Grain boundaries coupled to material flow 为No。Neighborhood选第一个,半径为1如图4所示
6. 位错密度常数设置 根据实际情况分别查找到对应材料各参数值,本次演示操作选取的值如图5所示 7. 再结晶设置 在再结晶界面选中Discontinous dynamic recrystallization (DRX)复选框,如图6所示,点击next 8. 形核状况设置 (1)在晶核形成界面1选中Function of threshold dislocation density and probability 单选按钮,如图7所示,然后next (2)在晶核形成条件界面2里,Critical dislocation density for DRX设为0.02,Probability of nucleation设为0.01,如图8所示,然后单击next。
“颗粒粒径分析方法”汇总大全
“颗粒粒径分析方法”汇总大全 来源:材料人2016-08-05 一、相关概念: 1、粒度与粒径:颗粒的大小称为粒度,一般颗粒的大小又以直径表示,故也称为粒径。 2、粒度分布:用一定方法反映出一系列不同粒径区间颗粒分别占试样总量的百分比称为粒度分布。 3、等效粒径:由于实际颗粒的形状通常为非球形的,难以直接用直径表示其大小,因此在颗粒粒度测试领域,对非球形颗粒,通常以等效粒径(一般简称粒径)来表征颗粒的粒径。等效粒径是指当一个颗粒的某一物理特性与同质球形颗粒相同或相近时,就用该球形颗粒的直径代表这个实际颗粒的直径。其中,根据不同的原理,等效粒径又分为以下几类:等效体积径、等效筛分径、等效沉速径、等效投影面积径。需注意的是基于不同物理原理的各种测试方法,对等效粒径的定义不同,因此各种测试方法得到的测量结果之间无直接的对比性。 4、颗粒大小分级习惯术语:纳米颗粒(1-100 nm),亚微米颗粒(0.1-1 μm),微粒、微粉(1-100 μm),细粒、细粉(100-1000 μm),粗粒(大于1 mm)。 5、平均径:表示颗粒平均大小的数据。根据不同的仪器所测量的粒度分布,平均粒径分、体积平均径、面积平均径、长度平均径、数量平均径等。 6、D50:也叫中位径或中值粒径,这是一个表示粒度大小的典型值,该值准确地将总体划分为二等份,也就是说有50%的颗粒超过此值,有50%的颗粒低于此值。如果一个样品的D50=5 μm,说明在组成该样品的所有粒径的颗粒中,大于5 μm的颗粒占50%,小于5 μm的颗粒也占50%。 7、最频粒径:是频率分布曲线的最高点对应的粒径值。 8、D97:D97指一个样品的累计粒度分布数达到97%时所对应的粒径。它的物理意义是粒径小于它的的颗粒占97%。这是一个被广泛应用的表示粉体粗端粒度指标的数据。 二、粒度测试的基本方法及其分析 激光法 激光法是通过一台激光散射的方法来测量悬浮液,乳液和粉末样品颗粒分布的多用途仪器。纳米型和微米型激光料度仪还可以通过安装的软件来分析颗粒的形状。现在已经成为颗粒测试的主流。 1、优点:(1)适用性广,既可测粉末状的颗粒,也可测悬浮液和乳浊液中的颗粒;(2)测试范围宽,国际标准ISO 13320 - 1 Particle Size Analysis 2 Laser Diffraction Meth 2 ods 2 Part 1: General Principles中规定激光衍射散射法的应用范围为0.1~3000 μm;(3)准确性高,重复性好;(4)测试速度快;(5)可进行在线测量。 2、缺点:不宜测量粒度分布很窄的样品,分辨率相对较低。 激光散射技术分类: 1、静态光散射法(即时间平均散射):测量散射光的空间分布规律采用米氏理论。测试的有效下限只能达到50纳米,对于更小的颗粒则无能为力。纳米颗粒测试必须采用“动态光散射”技术。 2、动态光散射法:研究散射光在某固定空间位置的强度随度时间变化的规律。原理基于ISO 13321分析颗粒粒度标准方法,即利用运动着的颗粒所产生的动态的散射光,通过光子相关光谱分析法分析PCS颗粒粒径。 按仪器接受的散射信号可以分为衍射法、角散射法、全散射法、光子相关光谱法,光子交叉相关光谱法(PCCS)等。其中以激光为光源的激光衍射散射式粒度仪(习惯上简称此类仪器为激光粒度仪)发展最为成熟,在颗粒测量技术中已经得到了普遍的采用。 激光粒度分析仪:
圆形提升盖板的设计计算.
圆形提升盖板的设计计算 1前言 平板是化工设备中最常见的部件。例如,各种容器的顶盖或顶板,设备的人孔盖板、法兰盖,施工中的管道试压盲板都属于此种类。其中圆形平板最为常见,本文提及的设备吊装用的圆形提升盖板就属于这一类。 石油化工装置中的一些特殊设备,如反应器、反应釜,由于其体积大、重量大、壁厚大,常用耐热合金钢制造,且经过整体热处理,所以在设计中往往不在壳体上布置吊耳,而是利用其顶部管口来进行吊装,提升盖板式吊耳设计正是为了满足这一要求而产生的。本文就有关的结构及强度计算进行论述。2圆形提升盖的结构形式 2.1结构如图1 提升盖的主要结构由法兰盖板和吊耳板组成,吊耳板可为单个也可使用二个。吊耳板与盖板间采用焊接形式。当板厚特别大时也可采用铸钢件,盖板与设备接口的连接采用法兰螺栓连接形式,可使用设备带来的螺栓。 为增加耳板的侧向刚度和耳板与盖板连接强度可在二者间设置肋板。 通常吊耳板用卡环及钢丝绳与吊装机械连接,故耳板尺寸与所用卡环应匹配。重型吊装盖板也可通过专用连接件与吊装机械连接。吊装盖板通常应随设备提供。 2.2提升盖的结构种类 按照提升盖板与设备管口的接触部位分类: a、不承受螺栓弯矩的盖板 此盖板与设备接口的接触部位仅为法兰螺栓圆部位见图2-1a、b。 b、承受螺栓弯矩的盖板 此盖板与设备接口的密封面部位相接触,因此螺栓预紧时产生的弯矩会叠加到盖板上(见图3)。这二类盖板在设计结构形式上有所差别,其力学模型不同,在设计计算中所用公式也不一样。 由于设备接口密封面往往高于法兰螺栓接圆面,设计盖板时应予以充分注意。
3圆形盖板计算的理论 3.1薄板理论基础 从设计观点看,板可分为厚、薄两种,厚板和薄板的理论基础和计算方法是不一样的,薄板的计算方法是厚板算法的一个特例,故掌握厚板理论完全可以解决问题。但厚板理论比较复杂,对于一般化工设备而言,大部属于薄板范围。 薄板理论又称为薄膜理论,其特点是只受拉力,不存在弯曲应力,该理论还有几点假设: 1)板的厚度较其它尺寸小得多。 2)中间面挠度比板厚小得多,即挠度很小。 3)中间面在弹性变形后仅有弯曲而不伸长。 4)原垂直于中间面的界面,变形后仍保持平面,且仍垂直于中间面。 3.2薄板理论应用条件 薄板理论是用小挠度理论假定推导的,因涉及到允许误差和计算精度,它的使用条件是: 1)薄板的定义是厚度小于其它两个尺寸的1/10时。 2)挠度小于1/5板厚时为小挠度。但从实用观点,既使挠度达到1/2板厚时,仍可作小挠度板计算。 3.3盖板计算的理论依据 吊装盖板的受力状态,基本上与压力容器的可拆卸平盖的形式相似,压力容器的平盖为均布荷载,而吊装盖板为局部载荷。 可拆卸平盖的计算方法在原压力容器设计规定中有两种,对于“中低压”部分是按薄圆平板公式计算,“高压”部分则按“巴赫公式”计算,二者计算结果存在着差别,但在82年版《设计规定》中已统一,计算公式的形式都采用薄圆平板的计算公式。 容器设计中可拆卸平盖计算时受力假设为: 1)均布载荷P作用在筒支的圆平板上。 2)螺栓载荷Wp作用在圆平板周边螺栓圆处。
晶粒度检验
《钢材质量检验》单元教学设计一、教案头
二、教学过程设计
三、讲义 1.金属的硬度试验 晶粒度检验 晶粒度是晶粒大小的量度,它是金属材料的重要显微组织参量。钢中晶粒度的检验,是借助金相显微镜来测定钢中的实际晶粒度和奥氏晶粒度。 实际晶粒度,就是从出厂钢材上截取试样所测得的晶粒大小。而奥氏晶粒度则是将钢加热到一定温度并保温足够时间后,钢中奥氏晶粒度大小。下面介绍奥氏晶粒度的显示和晶粒度的测定方法。 晶粒度的测定 在国家标准GB6394-86中规定测量晶粒度的方法有比较法、面积法和截点法等,生产检验中常用比较法。 1.比较法 比较法是在100倍显微镜下与标准评级图对比来评定晶粒度的。标准图是按单位面积内的平均晶粒数来分级的,晶粒度级别指数G和平均晶粒数N的关系为式中 N=2G+3 N-放大100倍时每1mm2面积内的晶粒数,晶粒越细,N越大,则G越大。 在GB6394-86中备有四个系列的标准评级图,包括I无孪晶晶粒,II有孪晶晶粒,III 有孪晶晶粒(深反差腐蚀),IV钢中奥氏体晶粒。图4-10是系列I的标准评级图。实际评定时应选用与被测晶粒形貌相似的标准评级图,否则将应引入视觉误差。当晶粒尺寸过细或过粗,在100倍下超过了标准评级图片所包括的范围,可改用在其他放大倍数下参照同样标准评定,再利用表查出材料的实际晶粒度。 评级时,一般在放大100倍数的显微镜下,在每个试样检验面上选择三个或三个以上具有代表性的视场,对照标准评级图进行评定。 若具有代表性的视场中,晶粒大小均匀,则用一个级别来表示该种晶粒。若试样中发现明显的晶粒不均匀现象,则应当计算不同级别晶粒在视场中各占面积的百分比,若占优势的晶粒不低于视场面积的90%时。则只记录一种晶粒的级别指数,否则应当同时记录两种晶粒度及它们所占的面积,如6级70%-4级30%。 比较法简单直观,适用于评定等轴晶粒的完全再结晶或铸态的材料。比较法精度较低,为了提高精度可把标准评级图画在透明纸上,再覆在毛玻璃上与实际组织进行比较。 四、训练任务
晶粒度测试与判定
1.晶粒度 晶粒大小的度量称为晶粒度。通常用长度、面积、体积或晶粒度级别数等不同方 晶粒大小的度量称为晶粒度通常用长度面积体积或晶粒度级别数等不同方 法评定或测定晶粒度大小。使用晶粒度级别数表示的晶粒度与测量方法和计量单 位无关。 2.实际晶粒度(如按照产品实际热处理条件进行渗碳淬回火后进行测试的晶粒度)实际晶粒度是指钢在具体热处理或热加工条件下所得到的奥氏体晶粒大小。实际 晶粒度基本上反映了钢件实际热处理时或热加工条件下所得到的晶粒大小,直接 影响钢冷却后所获得的产物的组织和性能平时所说的晶粒度如不作特别的说明 影响钢冷却后所获得的产物的组织和性能平时所说的晶粒度,如不作特别的说明,一般是指实际晶粒度。 3.本质晶粒度(如按照GB/T6394中渗碳法进行测试的晶粒度) 本质晶粒度是用以表明奥氏体晶粒长大倾向的晶粒度,是一种性能,并非指具体 的晶粒。根据奥氏仁晶粒长大倾向的不同,可将钢分为本质粗品粒钢和本质细晶 粒钢两类。就是这个材料的底子好不好,耐热处理晶粒不长大的能力好不好。 测定本质晶粒度的标准方法为:将钢加热到930℃±10℃,保温6h后测定奥氏体 晶粒大小,晶米度在1级~4级者为本质粗晶粒钢,晶粒度在5级~8级者为本质细 晶粒大小晶米度在级级者为本质粗晶粒钢晶粒度在级 晶粒钢。加热温度对奥氏体晶粒大小的影响见下图
一般情况下,本质细晶粒钢的晶粒长大倾向小,正常热处理后获得细小的实际晶粒,淬火温度范围较宽,生产上容易掌握,优质碳素钢和合金钢都是本质细晶粒钢。本质粗晶粒钢的晶粒长大倾向大,在生产中必须严格控制加热温度。以防过热晶粒粗化。值得注意的是加热温度超过930℃。本质细晶粒钢也可能得到很粗大的奥氏体晶粒。甚至比同温度下本质粗晶粒钢的晶粒还粗。 至比同温度下本质粗晶粒钢的晶粒还粗
内压薄壁壳体强度计算
第三章、 3—1 内压薄壁壳体强度计算 目的要求:使学生掌握内压圆筒内压球形壳体的强度计算,以及各类厚度的相互关 系。 重点难点:掌握由第一强度理论推出的内压圆筒,内压球形壳体的强度计算公式。 第三章 内压薄壁容皿 本章的任务就是在回转薄壁壳体应力分析的基础上,推导出内压薄壁容皿强度计公式。本章的压力容皿设计计算公式,各种参数制造要求以及检验标准均与GB150-1998《钢制压力容皿》保持一致。 第一节 压内薄壁壳体强度计算 一、 内压圆筒 为了保证圆筒受压后不破裂,[根据第一强度理论]应使筒体上最大应力,即环向应力2σ小于等于材料在设计温度下的许用应力[]t σ 用公式表达:2[]2t P D σσδ =≤g ,其中P-设计压力。 1)中径0 () 2i D D + 此外还应考虑到,筒体在焊接的过程中,对焊金属组织的影响以及焊接缺陷(夹渣、气孔、未焊透等)影响缝焊的强度(使整本强度降低),所以将钢板的许用应力乘以一个小于1的焊接接头系数,以弥补焊接可能出现的强度削弱,故 2[]2t P D σσδ=≤g :[]2t P D σ?δ≤g g 此外,工艺计算时通常以i D 做为基本尺寸,故将i D D δ=+代入上式: 则 () []2t i P D δσ?δ +≤g 可解出δ,同时根据GB150-1998规定,确定厚度时的压力用计算压力c p 代替。 最终内压薄壁圆筒体的计算厚度δ: 2[]C i t C P D P δσ?= -g 适用:0.4[]t C P σ≤ 考虑到介质时皿壁的腐蚀,确定钢板厚度时,再加上腐蚀裕量: 2C d δδ+=——圆筒的设计厚度 再考虑到钢板供货时的厚度偏差,将设计厚度加上厚度负偏差,再向上圆整三规格
jade分析物相与晶胞参数和晶粒尺寸计算过程
《无极材料测试技术》课程作业 对编号 01N2009534 的样品 XRD 测试数据进行物相分析,并计算其平 均晶粒尺寸大小与晶胞参数。 1. 物相分析过程 使用 MDI Jade5.0 软件对样品 XRD 测试数据进行分析,以定性分析样品的物相。 1.1. 数据的导入 将测试得到的 XRD 测试数据文件 01N2009534.txt 直接拖动到 Jade 软 件图标上,导入数据,得到样品 XRD 衍射图(图 1-1)。 图 1-1 数据导入 Jade5.0 后得到的 XRD 图 1.2. 初步物相检索 右键点击 键,弹出检索对话框,设定初步检索条件:选择所有类 型的数据库;检索主物相( Major Phase );不使用限定化学元素检索( Use Chemistry 前方框不打钩)(如图 1-2 所示)。点击“ OK ”开始检索,得到的检索结果见图 1-3。 从初步检索结果可以看出,最可能的物相有四个: 5 8 323(图 1-3 )、 CaB 6 O 10 · 5H 2 O ( 图 1-4a )、 CaB O (OH)B(OH) (H O) 2.62 Al 9.8 Si 26.2 O 72 H 4.56(图 1-4b )和 C 20 20 16 8 4(图 1-4c )。其中前 Ca H N O S Th 三个均为无机物,第四个为有机金属化合物。
从结果分析,由图 1-4b、c 中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图 1-3 以及图 1-4a 中可以看出,两种 物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为 CaB5O8(OH)B(OH) 3(H 2O) 3或 CaB6 O10·5H2O 或者两者的混合物。 图 1-2 初步物相检索条件设定 图 1-3 经过初步检索得到的检索结果
对不同颗粒尺寸采用不同的测量方法
对不同颗粒尺寸采用不同的测量方法 作者:Stephen Ball,,马尔文仪器产品营销经理 本文中,马尔文仪器产品营销经理Stephen Ball向您介绍生物制药中蛋白质团聚物的一些测量技术。 随着生物分子在许多制药公司药物开发途径中所占据的比例越来越大,人们越来越关心相关开发、生产与监管方面难题的解决。由于药物的潜在免疫原性是生产商和监管者都十分关心的要素,因此如何定义生物药品的纯度与效力要比那些小分子药物复杂得多。这反过来突显了业界对高质量分析工具的迫切需要——希望它们能有助于全面表征出生物药物颗粒和团聚物,同时对药物内在颗粒与污染物的理化特性表征也越来越重视。 测量的用处何在? 药物分子从发现走向早期配方是十分关键的一个步骤。分子的理化特性是药物配方与给药的决定因素;药物分子的理化特性确定得越早,就能获得越大的经济效益——无论是为了确定上述步骤成功的可能性,还是尽早避免可能的失败。就这一点而言,比较理想的是能够在非常少量的样品上进行一系列非破坏性试验,并且把更多的精力放在改善可能用到的测试过程中。 从药物分子理化特性表征到药物开发过程直至最终的成品测试,发现和检测蛋白质团聚物,是药物开发最重要的步骤,因为理解药物分子这方面的行为对于药物产品的配制、稳定性和安全性来说都是至关重要的。蛋白质结构是通过范德华力、氢键、二硫键和疏水作用的结合来保持的,环境条件的改变可能会影响其中的一些或者全部相互作用力——其结果可能会引发团聚物非正常的折叠或者对于溶解性造成负面影响。在此过程中,蛋白质的活性常常会消失,也有可能很多团聚物会发展出免疫原性,从而对最终治疗药物产品的有效性和安全性造成显著影响。 当前的监管预期是,对团聚物和大小范围在“0.2- 2”微米级别1的不溶性微粒进行表征。大小超过几微米的团聚物可以用视觉方法来进行表征。小于这个尺寸的,可以采用动态光散射(DLS) 法和体积排除色谱 (SEC) 法等成熟的分析技术对蛋白质凝聚物进行表征。图1列出了上述方法所采用的技术和测量范围。共振质量测量法 (RMM)是最新发展的技术现在也被用来检测和统计50 nm到5μm这一至关重要尺寸范围内的不溶性微粒,并对它们的浮力质量、净质量和粒径大小进行可靠的测量。共振质量测量法为不溶性微粒和亚微米级凝聚物提供了测量窗口。 动态光散射法的使用 动态光散射法测量迅速,属于非侵入性的测量方式,特别适合在药物试剂开发的早期阶段筛选蛋白质。
deform中晶粒模拟
晶粒模拟 1.输入变形主要文件 2.输入与晶粒有关的材料参数 3.输入最初的晶粒变量 4.运行模拟 5.准备及运行空冷模拟 6. 准备及运行水中淬火模拟 7.后处理 8.改变条件 介绍 本章的目的是介绍如何采用DEFORM2D晶粒模拟模拟锻 造过程及热处理过程中微观组织的变化。 再结晶度及平均晶粒尺寸是使用者最关心的参数,该模型中共有16中晶粒变量,他们都放在数据库中。 静态再结晶、中间动态再结晶、动态再结晶的演化机理和结晶成长都在模型中被计算。在每一个时间步里,基于时间、温度、应力、应力速率、演化历史,变形机制被定义,晶粒的变化被计算和更新。关于该模拟完整的解释在用户文档中有。 注意: 1)由于锻造过程的复杂性,对动态再结晶的同步模拟几乎是不可能的。实际上动态再结晶的计算是在变形过程之后。中间动态
再结晶,动态再结晶也是如此。这就是说,用户将看不到任何的 结果除非一个非变形的模拟(例如:热处理)跟在一个变形模拟的后面。 2)要完成一个完整的晶粒变化模拟,用户必须确定一个完整的热处理过程。特别是坯料必须在模拟结束时彻底的冷却。 问题摘要 空冷水中淬火是一个既简单又让人头疼的过程,该问题 使用SI单位,轴对称。材料IN718,模具材料H13钢。 1.输入变形主要文件 做一个工作路径,打开DEFORM 2D,用Problem ID GRAIN_LAB, 打开前处理,装载KEY文件UPSET.KEY. 这个KEY文件包含了该模拟的所有信息。 2.输入与晶粒有关的材料参数 点击模拟控制按纽,激活“晶粒”,到材料中选择IN718,点击晶粒窗口,窗口显示如下: 激活meta-dynamic、grain growth,不激活其他俩个,输入以下数据到相应的矩阵。 最高应力 应变速率极限 中间动态再结晶动力 中间动态再结晶晶粒尺寸
油中颗粒数及尺寸分布测量方法
中华人民共和国能源部标准 SD313—89 油中颗粒数及尺寸分布测量方法 (自动颗粒计数仪法) 中华人民共和国能源部1989-03-27批准 1989-10-01实施 本方法采用自动颗粒计数仪测定每100mL油中所含颗粒数量及尺寸分布,测量颗粒尺寸范围5~150μm(更换传感器可以扩大量程)。适合于测定变压器油、汽轮机油等油品的颗粒污染度。 1仪器及材料 1.1自动颗粒计数仪根据遮光原理工作,需定期校准。 1.2传感器与自动颗粒计数仪配套使用,能测定粒径约150μm的颗粒。 1.3超声波清洗器最小功率50W。 1.4秒表或计时器精确度为0.1s。 1.5真空泵真空度不小于86kPa。 1.6过滤装置供过滤清洁液使用。 1.7微孔滤膜孔径为0.8μm、0.45μm和0.15μm。 1.8取样瓶250mL玻璃瓶(医用输液瓶),具塞和塑料薄膜衬垫。 1.9AC粉尘校准液AC粉尘为非球形颗粒,校准液一般由仪器生产厂提供,也可按国际标准ISO4402方法配制。 1.10标准胶球校准液一般由仪器生产厂提供,也可按美国宇航标准ARP1192A方法配制。 1.11异丙醇化学纯。 1.12石油醚化学纯,沸程60~90℃。 1.13甲苯(或二甲苯)化学纯。 1.14去离子水或蒸馏水。 2清洁液的制备 异丙醇、石油醚、甲苯和蒸馏水等可依次经过不同孔径的滤膜过滤制得。供清洗仪器及稀释样品使用的清洁液,每100mL中粒径大于5μm的颗粒不多于100粒;供检验取样瓶用的清洁液,每100mL中粒径大于5μm的颗粒不多于50粒。 3取样 3.1取样瓶的清洗及检验 3.1.1取样瓶经过自来水和蒸馏水清洗后,再用清洁水清洗,瓶盖和薄膜衬垫也要用清洁液清洗。 3.1.2检验清洗干净后,向瓶中注入总容积为45%~55%的清洁液,垫上薄膜,盖上瓶盖后充分摇动。用自动颗粒计数仪测定每100mL液体中粒径大于5μm的颗粒数应不多于100
Jade 是如何计算晶粒尺寸的
Jade 是如何计算晶粒尺寸的? 不止10次有人问到这个问题,让我有兴趣去了解。看了看这个软件的帮助,也没有得到答案。只好一种一种方法去试,好象还真是得到了解答。今天,把它写出来供大家验证。 Jade 按照谢乐公式来计算。 θ βλcos k D = λ 是辐射的波长,按K α1的波长计算,如铜靶,则λ=0.154056nm 。 D 就是晶块尺寸,单位可以是纳米,与波长λ的单位相同。 k 是一个参数,可以取0.89,0.95或者1,一般人都愿意取1。但是,软件是按0.89计算的。 θ是半衍射角,单位可以是度或者弧度,只要你能正确计算出它的余弦就可以。 β是衍射峰的加宽。一般按两种方法来计算,即b B ?=β,22b B ?=β一般人愿意用b B ?=β。但是,Jade 却用后者。确实,一些教科书中都提到,后者更符合实际情况。 这里的B 就是FWHM ,即样品的衍射峰宽,b 则是仪器宽度。 好了。让大家来看看我的试验过程。 有这么一个衍射峰,我们先来做拟合:
通过Report----peak profile report菜单,查看到拟合的结果: 通过菜单Edit-----Preferences,可看到下面的窗口:
单击View FWHM Curve,你看到: 你可能看到的不一样,这是因为你没有做仪器校正,而使用了软件自带的某个“标样”,如Constant FWHM。这里看到的是我在07年12月19日做的硅标数据。 移动你的鼠标,并定位于116°处,你可看到FWHM=0.140°。这就是仪器宽度,即b。
在这个窗口中,你还看到了仪器波长是 1.54056埃,即0.145056nm。 怎么样?把这些数据代入到公式,得到14.40902nm。 这里讲的是单峰处理时的晶块尺寸。要注意,除非你的样品是分散单体纳米晶,否则,这个数据是不可信的。 关于晶块尺寸计算与微观应变更详细的解释,请访问我的QQ空间,也许会有些帮助。
Image J分析粒径尺寸及分布
Morphological study: focal contacts formation For quantification of vinculin positive contacts areas, we used the freeware image analysis ImageJ (NIH, https://www.wendangku.net/doc/287971117.html,/ij/). We opened the raw image, converted it to an 8-bit file, and used the unsharp mask feature (settings 1:0.2) before removing the image background (rolling ball radius 10). After smoothing, the resulting image, which appears similar to the original photomicrograph but with minimal background, was then converted to a binary image by setting a threshold. Threshold values were determined empirically by selecting a setting, which gave the most accurate binary image for a subset of randomly selected photomicrographs with varying peptides densities. The cell area was determined by manual delineation on raw fluorescent images, total contact area and mean contact area per cell were calculated by ‘‘analyse particules’’ in Image J, contacts smaller than 3 pixels were not taken into account. 首先用工具栏里的直线工具,沿SEM、TEM的标尺(bar)拉出一条等长的直线,在菜单栏里找到measure点击,得到标尺对应的长度数据(弹出的数据栏);接着用同样的步骤测量你的particles;最后导出你的数据,放到excel、origin里处理就行了~ 测量的值是相对值,要利用bar的相对值和实际值进行换算~乘下除下就行了~ 首先要在measur 里面选择spatial calibration,将这里面标尺与自己图片里面的标尺对应,最后选择measurements,features里面选择直线。自己在图片里面画线,用长度除以晶粒数就ok了
管道压力试验封头型式及厚度的确定
长输管道压力试验封头型式及厚度的确定 郭明万 摘要:根据长输管道的材质和压力等级,匹配常用的压力容器用钢板作为管道压力试验封头用材料,按压力容器的方法确定封头的结构型式和厚度。 关键词:压力试验;封头;厚度 符号说明 δ——计算厚度,mm; P ——计算压力,MPa;等于设计压力与压力试验管段液位高差静压力之和; c ——封头内直径,mm; D i [σ]t——设计温度下材料的许用应力,MPa; φ——焊接接头系数,采用整板料取1; α——圆锥半顶角,(°); 压力试验是管道施工涉及人身和财产安全的关键工序,在管道设计规范、施工规范中均未对管道压力试验的封头型式、材质与厚度作出相应的规定,施工单位一般根据经验和材料的实际情况确定,存在着较大的安全风险。但压力管道(最大直径φ1219mm,最高设计压力10MPa)与压力容器(最大直径超过φ5000mm,最高设计压力大于100MPa)同属承压类特种设备,把管道等同于筒体很长的压力容器,管道压力试验与压力容器的压力试验就是完全相同的,因此,用压力容器的方法确定长输管道试压封头是满足管道要求的。管道压力试验的封头型式、材质与厚度可以根据压力容器的基本要求和计算方法确定。
1 封头型式的确定 压力容器用封头根据几何形状的不同,一般分为球形封头、椭圆封头、碟形封头、锥形封头、平盖等。以峰值应力和截面突变情况为依据,优先选用球形封头,其它封头依次次之,平盖的受力状况最差,截面突变最大。 1.1球形封头 球形封头截面形状为半球形,球形封头没有相应的专业制造标准,到目前为止,一般按照GB150进行设计计算,参照JB/T4746制造,根据需要,封头直边可有可无,供需双方协商确定。由于截面突变最小,其受力状况最好,在同等条件下所需的金属厚度最小,其厚度计算公式为: δ= P c D i 4[σ]tφ-P c 但由于封头深度较大,加工难度相对较大,且考虑到与管道(筒体)等厚度焊接的因素,从经济适用出发,球形封头一般用于压力较高的场合才能体现其受力状况佳、用料厚度较小的优势。建议设计压力≥8.0MPa的管道采用球形封头作为试压封头。 1.2椭圆封头(本文指标准椭圆封头) 椭圆封头截面形状为半椭圆形,按GB150进行设计计算,按JB/T4746制造加工。其截面突变和受力状况仅次于球形封头,加工深度较小,使用最普遍,标准椭圆封头厚度计算公式为: δ= P c D i 2[σ]tφ-0.5P c 建议设计压力<8.0MPa的管道采用标准椭圆封头作为试压封头。 1.3碟形封头 使用较少,不采用。
Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸)
Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) Scherrer公式计算晶粒尺寸(XRD数据计算晶粒尺寸) 根据X射线衍射理论,在晶粒尺寸小于100nm时,随晶粒尺寸的变小衍射峰宽化变得显著,考虑样品的吸收效应及结构对衍射线型的影响,样品晶粒尺寸可以用Debye-Scherrer公式计算。 Scherrer公式:Dhkl=kλ/βcosθ 其中,Dhkl为沿垂直于晶面(hkl)方向的晶粒直径,k为Scherrer 常数(通常为0.89),λ为入射X射线波长(Cuka 波长为0.15406nm,Cuka1 波长为0.15418nm。),θ为布拉格衍射角(°),β为衍射峰的半高峰宽(rad)。 但是在实际操作中如何从一张普通的XRD图谱中获得上述的参数来计算晶粒尺寸还存在以下问题: 1) 首先,用XRD计算晶粒尺寸必须扣除仪器宽化和应力宽化影响。如何扣除仪器宽化和应力宽化影响?在什么情况下,可以简化这一步骤? 答:在晶粒尺寸小于100nm时,应力引起的宽化与晶粒尺度引起的宽化相比,可以忽略。此时,Scherrer公式适用。但晶粒尺寸大到一定程度时,应力引起的宽化比较显著,此时必须考虑引力引起的宽化, Scherrer公式不再适用。
2) 通常获得的XRD数据是由Kα线计算得到的。此时,需要Kα1 和Kα2必须扣除一个,如果没扣除,肯定不准确。 3) 扫描速度也有影响,要尽可能慢。一般2°/min。 4)一个样品可能有很多衍射峰,是计算每个衍射峰对应晶粒尺寸后 平均?还是有其它处理原则? 答:通常应当计算每个衍射峰晶粒尺寸后进行平均。当然只有一两 峰的时候,就没有必要强求了! 5) 有的XRD数据中给出了width值,是不是半高宽度的值?能不能 直接代入上面公式吗?如果不能,如何根据XRD图谱获得半峰宽? TOP 20 β为衍射峰的半高峰宽时,k=0.89 β为衍射峰的积分宽度时,k=1.0。其中积分宽度=衍射峰面积积分/峰高 如何获得单色Kα1: 1)硬件滤掉Kβ:K系射线又可以细分为Kα(L层电子填充)和Kβ(M层电 子填充)两种波长略有差异的两种射线。而X射线衍射仪要求使用单色X射线,因此,需要在XRD实验时把后者除掉。 a). 传统的方法是在光路上加入一个滤波片(如Ni)。 b).现在一般使用铜靶,在光路上增加一个石墨晶体单色器来去除Kβ射线。通常的做法是在衍射线的光路上,安装弯曲晶体单色器。石墨单晶体单色器是一块磨成弯曲面的石墨单晶体。由试样衍射产生的衍射线(称为一次衍射)经单色器时,通过调整单晶体的方位使它的某个高反射本领晶面与一次衍射线的夹角刚好等于该晶面对一次衍射的Kα辐射的布拉格角。单色器可以去除衍射背底,也可以去除Kβ射线的干扰。这样,由单晶体衍射后发出的二次衍射线就是纯净的与试样衍射对应的Kα衍射线。 2) 软件分离Kα2:Kα辐射还可以细分为Kα1和Kα2两种波长差很小的辐射。由于它们的波长差很小,无法通过硬件的方法来消除其中任何一种,因此,只有