一篇关于NMR的理论计算的文章

OPBE:A promising density functional for the calculation
of nuclear shielding constants
Ying Zhang a ,Anan Wu a ,Xin Xu
a,*
,Yijing Yan
b
a
State Key Laboratory of Physical Chemistry of Solid Surfaces,Center for Theoretical Chemistry,College for Chemistry and Chemical
Engineering,Xiamen University,YanWu Road,Xiamen,Fujian 361005,China
b
Department of Chemistry,Hong Kong University of Science and Technology,Kowloon,Hong Kong
Received 1January 2006;in ?nal form 26January 2006
Abstract
We investigate the performance for nuclear magnetic constant calculations for a selective set of density functional methods (B3LYP,PBE0,BLYP,PBEPBE,OLYP and OPBE).The testing set includes the 13C,15N,17O and 19F magnetic shieldings and chemical shifts of 23molecules with 64comparisons altogether.The results are compared and contrasted to the experimental gas phase data.We ?nd that the OPBE exchange-correlation functional performs remarkably well for the whole set of the testing system,rendering OPBE the best GGA (generalized gradient approximation)functional,determined from energy criteria,for the prediction of nuclear magnetic constants.ó2006Elsevier B.V.All rights reserved.
1.Introduction
There is a great leap forward in the calculations of NMR properties in the past few decades [1,2].The calcula-tions from the ?rst principles have reached the point of providing quantitative agreement with experiment in some cases.For small,isolated systems,accurate results for the NMR chemical shifts (d e )may be achieved by using wave-function based ab initio approaches such as MP2[3],MP3[4],CCSD [5],CCSD(T)[6],and MC-SCF [7],etc.to take into account the correlation e?ects.However,such calcula-tions are computationally demanding,rendering them-selves impractical for larger molecules.
Density functional theory (DFT)o?ers an alternative way to treat the correlation e?ects more e?ciently [8–17].As its computational cost is in the same order of the Har-tree–Fock (HF)method,DFT is applicable to much larger systems than the correlated ab initio methods.Even though there exist some pitfalls for the conventional DFT meth-ods,for example,the current density dependency is omitted [14],and it is not free from self-interaction error [12],it is
not unreasonable to expect that a conventional DFT can lead to NMR properties with reasonable accuracy,judged on its great success in predicting various molecular proper-ties such as polarizibility,vibrational frequency,etc.[2].In fact,there are some pioneer works investigating the perfor-mance for DFT prediction of NMR properties.Taking the experimental chemical shifts (d 0)as reference,Cheeseman et al.claimed that DFT like BPW91,BLYP,B3PW91,or B3LYP statistically shows an improvement over the HF method,although DFT is still way behind the MP2method in accuracy [9].Adamo and Barone,based on a compari-son between their calculated absolute shieldings (r e )with the experimental values (r 0),concluded that the hybrid functional PBE0(also known as PBE1PBE)is able to deli-ver competitive results with respect to the MP2method [10].Nevertheless,Pulay and Magyarfalvi took the abso-lute shieldings (r e )obtained at the CCSD(T)level as refer-ence,giving a di?erent conclusion that the commonly used density functionals such as BLYP and B3LYP are signi?-cantly inferior to HF for general organic molecules,because the DFT methods always underestimate shieldings [11].Handy et al.concluded that exchange-correlation functionals determined from energy criteria are not optimal for nuclear shielding predictions;and that DFT is not yet
0009-2614/$-see front matter ó2006Elsevier B.V.All rights reserved.doi:10.1016/j.cplett.2006.01.095
*
Corresponding author.Fax:+865922183047.E-mail address:xinxu@https://www.wendangku.net/doc/8a11604821.html, (X.Xu).
https://www.wendangku.net/doc/8a11604821.html,/locate/cplett
competitive with ab initio correlated calculations for the prediction of the NMR shielding constants [8].
Unlike wavefunction based ab initio approaches,there lacks a systematic way of improving the quality of the results for an approximate DFT.The only recourse is to try a di?erent approximate functional,when the calculated properties fail to follow the experimental trends.In this contribution,we propose and present a validation of OPBE for the calculations of the nuclear magnetic constants.The results suggest that OPBE has promise as a widely applica-ble scheme for the prediction of NMR properties,yielding results competitive to or even better than the https://www.wendangku.net/doc/8a11604821.html,putational details
In this work,we have examined four pure GGA (gener-alized gradient approximation)functionals,namely,BLYP,PBEPBE,OLYP and OPBE,combining three exchange functionals with two correlation functionals.These included the commonly used exchange functionals such as Becke88[18]and PBE [19]and the popular corre-lation functionals such as LYP [20]and PBE [19].Espe-cially,we have tested the newly-developed exchange functional,OPTX [21],whose parameters were optimized by ?tting to the unrestricted HF energies of the ?rst-and second-row atoms.Hybrid functionals have distinguished themselves as the method of choice for the calculations of various molecular properties,hence we have also examined two hybrid functionals,B3LYP [22]and PBE0[10].For comparison,we have also included two ab initio methods,the restricted Hartree–Fock (RHF)and MP2.
In all NMR calculations,we used the gauge including atomic orbital (GIAO)method [23],as implemented in the G AUSSIAN 03program suite [24].We adopted the 6-311+G(2d,p)basis set as recommended by Cheeseman et al.[9].As for the molecular geometry,we pragmatically
choose to use the optimized one,obtained by using the same functional and basis set as those applied to the NMR calculation.
A set of 23molecules has been employed as a testing set in the present work [3–6].It contains molecules with single,double,and triple bonds,and systems requiring multiple determinants for proper descriptions;while reliable experi-mental NMR data are available.In this work,we take the measured gas phase NMR data (r 0,d 0)as reference,aiming to examine the performance of these functionals from a practical point of view.3.Results and discussion 3.1.Geometric parameters
Geometric optimization is now routinely applied in computational chemistry.The geometry obtained is the equilibrium geometry,r e ,with which we are going to calcu-late the nuclear magnetic constants.Nevertheless,it is pretty di?cult to determine experimentally the magnetic properties of a single non-vibrating molecule at a ?xed geometry;and there is a lack of a complete collection of experimentally determined r e [25,26].
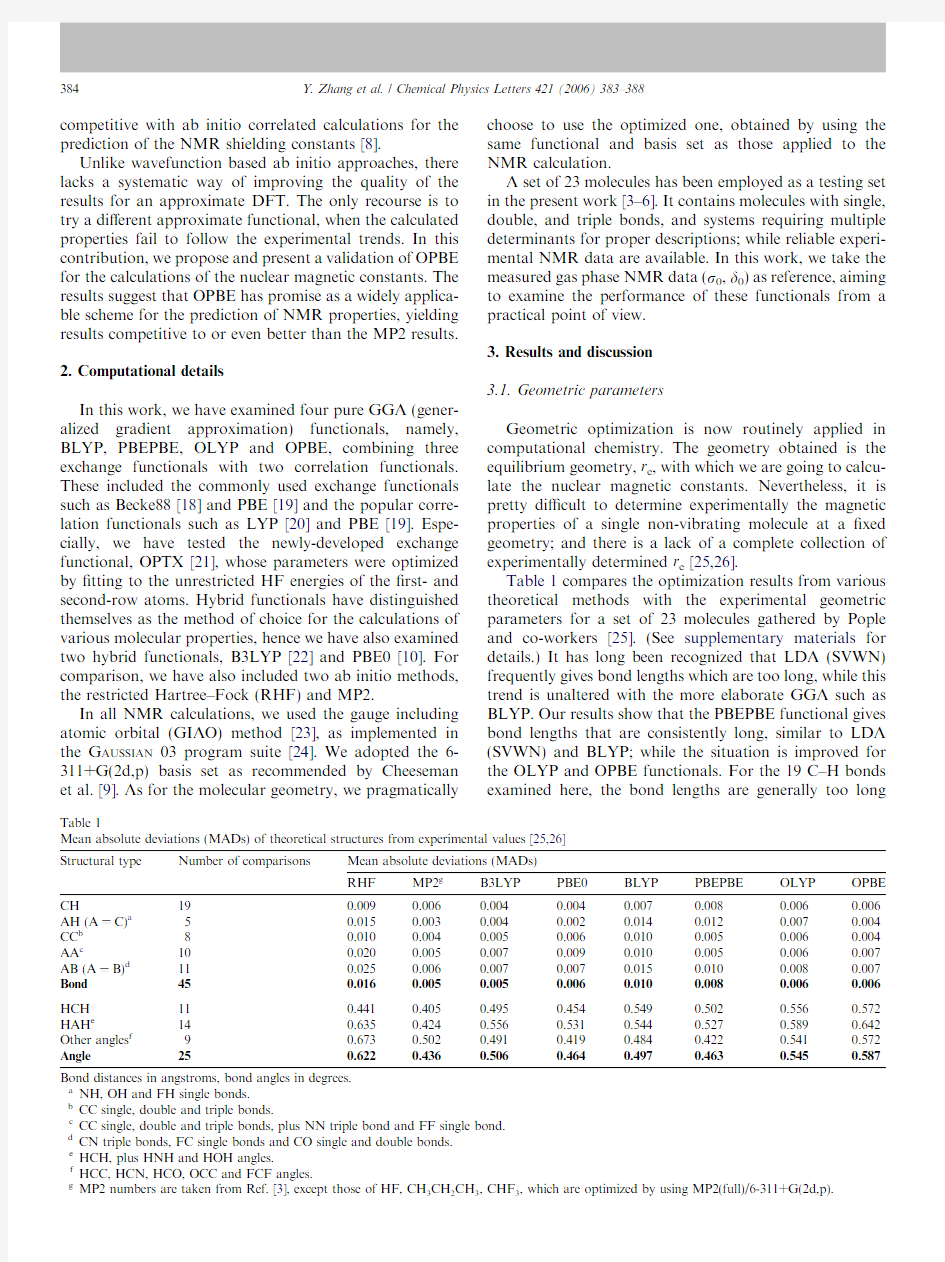
Table 1compares the optimization results from various theoretical methods with the experimental geometric parameters for a set of 23molecules gathered by Pople and co-workers [25].(See supplementary materials for details.)It has long been recognized that LDA (SVWN)frequently gives bond lengths which are too long,while this trend is unaltered with the more elaborate GGA such as BLYP.Our results show that the PBEPBE functional gives bond lengths that are consistently long,similar to LDA (SVWN)and BLYP;while the situation is improved for the OLYP and OPBE functionals.For the 19C–H bonds examined here,the bond lengths are generally too long
Table 1
Mean absolute deviations (MADs)of theoretical structures from experimental values [25,26]Structural type Number of comparisons Mean absolute deviations (MADs)RHF MP2g B3LYP PBE0BLYP PBEPBE OLYP OPBE CH
190.0090.0060.0040.0040.0070.0080.0060.006AH (A ?C)a 50.0150.0030.0040.0020.0140.0120.0070.004CC b 80.0100.0040.0050.0060.0100.0050.0060.004AA c
100.0200.0050.0070.0090.0100.0050.0060.007AB (A ?B)d 110.0250.0060.0070.0070.0150.0100.0080.007Bond 450.0160.0050.0050.0060.0100.0080.0060.006HCH 110.4410.4050.4950.4540.5490.5020.5560.572HAH e
140.6350.4240.5560.5310.5440.5270.5890.642Other angles f 90.6730.5020.4910.4190.4840.4220.5410.572Angle
25
0.622
0.436
0.506
0.464
0.497
0.463
0.545
0.587
Bond distances in angstroms,bond angles in degrees.a
NH,OH and FH single bonds.b
CC single,double and triple bonds.c
CC single,double and triple bonds,plus NN triple bond and FF single bond.d
CN triple bonds,FC single bonds and CO single and double bonds.e
HCH,plus HNH and HOH angles.f
HCC,HCN,HCO,OCC and FCF angles.g
MP2numbers are taken from Ref.[3],except those of HF,CH 3CH 2CH 3,CHF 3,which are optimized by using MP2(full)/6-311+G(2d,p).
384
Y.Zhang et al./Chemical Physics Letters 421(2006)383–388
by0.008(PBEPBE),and0.007A?(BLYP);while OLYP and OPBE lead to a mean absolute deviation(MAD)of 0.006A?,comparable to that of MP2.The hybrid function-als B3LYP and PBE0give the best results with MAD of only0.004A?.For the X–H bonds other than C–H,the sit-uation is worse for BLYP and PBEPBE,while it remains to be good for OLYP,OPBE,B3LYP and PBE0,being com-petitive to MP2.Except BLYP,all functionals give a simi-lar MAD to that of MP2for CC single,double and triple bonds.This is still true when di?cult cases,such as N2 and F2,are included for comparison.For bonds between non-hydrogen heteroatoms(A–B),the MADs are0.015 (BLYP),0.010(PBEPBE),0.008(OLYP)and0.007A?(B3LYP,PBE0and OPBE).The corresponding data for RHF and MP2are0.025and0.006A?,respectively.
We have also examined23bond angles.All functionals lead to bond angles which are always too small.The devi-ation from the experiment is around0.5°.
Based on the data in Table1,we?nd that the MADs for the prediction of bond lengths follow the order of MP2,B3LYP(0.005A?) 3.2.Magnetic shieldings First,let us examine the performance of di?erent meth-ods for the prediction of absolute magnetic shielding of 13C.We calculate the mean absolute deviations from the experiments.This set of molecules includes22comparisons for carbon nucleus in various environments(Table2). Take the experimental gas phase data(r0)as reference, it is seen that all the DFT methods underestimate the13C shieldings and the performance of some commonly used DFT methods such as BLYP(MAD=14.0)is signi?cantly inferior to RHF(MAD=7.0ppm).This is consistent with the conclusion of Pulay[11]and others[8].Indeed,other functionals like PBEPBE and also hybrid B3LYP perform no better than RHF.(See supplementary materials for details). It is apparent that the MP2method systematically over-estimates the13C shieldings and leads to a MAD of 10.0ppm;while MAD of the hybrid functional PBE0is only3.3ppm,signi?cantly lower than that of the MP2 method.This is in line with the?nding of the previous investigation of Adamo and Barone[10]. Our work revealed that some newly-developed GGA functionals,OLYP and OPBE,which used OPTX as the exchange functional,exhibit remarkably good perfor-mance.OPBE is the best method investigated in our work, leading to a MAD of only2.1ppm.Signi?cantly,we see that there are many cases where OPBE greatly surpasses the MP2(e.g.MAD(OPBE/MP2)=0.28/9.3for CH3OH, 0.39/8.5for CH4,0.16/9.9for CH3F,1.13/12.5for C2H4, etc.See supplementary materials for details.) It is a challenge to give an accurate prediction of mag-netic shieldings for heteroatoms of15N,17O,19F.Since the correlation e?ects play an important role,MAD from the RHF method is increased dramatically to33.0ppm; while that of MP2is23.1ppm.For this set of12compar-isons,all DFT methods are superior to the RHF method. In fact,except BLYP,the other functionals perform even better than MP2.B3LYP and PBE0lead to MADs of 10.8and13.6ppm,respectively.OLYP and OPBE are the best performers,giving MADs of10.6and9.9ppm, respectively. Table2 Mean absolute deviations(MADs)of the theoretical magnetic constants from the experimental values RHF MP2B3LYP PBE0BLYP PBE OLYP OPBE Magnetic shieldings r MAD(C)a7.010.08.8 3.314.08.9 4.4 2.1 MAD(O,F,N)b33.023.110.813.626.817.410.69.9 MAD(tot)16.214.69.5 6.918.511.9 6.6 4.9 Maximum error253.954.8à20.488.5à115.5à46.7à33.243.1 (F2)(F2)(CH F3)(F2)(F2)(F2)(CH F3)(F2) Chemical shifts d MAD(C)a 6.5 2.7 3.9 2.6 6.8 4.9 2.9 2.3 MAD(O,F,N)b35.610.011.117.924.116.710.312.6 MAD(tot)15.2 4.9 6.17.211.58.4 5.1 5.4 Maximum errorà242.3à39.2à19.3à86.6106.641.130.1à43.4 (F2)(F2)(F2)(F2)(F2)(F2)(CH F3)(F2) The maximum error(theoryàexperiment)for each method is also included.Numbers in bold faces represent the best two methods in each entry. a Testing set for13C includes C H 4 ,C2H2,C2H4,C2H6,H2CC CH2,C H3C H2CH3,C6H6,C H3F,C HF3,C F4,C O2,C O,H2C O,C H3OH C H3C OCH3, H C N,C H3C N,C H3NH2.Experimental values for13C are taken from Refs.[9,13,28]. b Testing set for15N,17O,19F includes H 2 O,N H3,H F,F2,CH3F,CH F3,C O2,C O,N2,HC N,CH3C N,CH3N H2.Experimental values for15N are taken Refs.[9,13,29],17O values from Refs.[30,31],and19F values from Refs.[9,13,32]. Y.Zhang et al./Chemical Physics Letters421(2006)383–388385 The performance of the OLYP functional has been investigated before[11,15].Using a di?erent testing set, Cohen et al.obtained a MAD of33.7ppm.This is compa- rable to that of HCTH(35.5ppm),which was claimed to be the best GGA functional,optimized based on energy criteria,for the NMR prediction[8].Cohen et al.showed that BLYP led to a MAD of49.5ppm;while B3LYP gave a MAD of64.5ppm,thus are signi?cantly inferior to OLYP.Noteworthily,KT2[17],a functional speci?cally optimized for NMR calculation lead to a MAD of only 16.2ppm[15]. There are some di?cult cases(e.g.F2,CHF3),which are related to19F(see supplementary materials for details).For F2,errors(theoryàexperiment)areà17.5(B3LYP),à88.5 (PBE0),à43.1(OPBE),18.7(OLYP),115.5(BLYP),46.7 (PBEPBE),à253.9(RHF),andà54.8(MP2).The dra- matic errors for RHF,PBE0and BLYP may be traced to the errors in predicting the F–F bond distance (MAD=0.087(RHF),0.038(PBE0)and0.020A? (BLYP)).RHF optimization leads to the F–F distance of 1.330A?,giving the NMR19F shielding of21.12ppm; whereas if the experimental F–F distance of1.417A?[25] is used,the NMR shielding by RHF isà184.92ppm. The increase of the F–F distance by0.087A?results in a decrease of the calculated19F shielding by206ppm.Simi- larly,PBE0gives the optimized F–F distance of1.379A? and the NMR19F shielding ofà144.29ppm;increasing the F–F distance to the experimental one brings a decreas- ing of shielding by109ppm.On the other hand,optimiza- tion by BLYP leads to an elongated F–F distance (1.437A?),this is accompanied by a too deshielded nuclear with shielding ofà348.26ppm;decreasing the F–F dis- tance to the experimental one increases the shielding by 65ppm.Thus we see that the incorrectly calculated equilib- rium geometry has a profound e?ect on the prediction of the NMR shieldings. Overall,we?nd that the MADs for the prediction of magnetic shieldings follow OPBE(4.9) PBE0(6.9) (14.6) 3.3.Chemical shifts Table2summarizes the statistical data for13C,15N,17O and19F chemical shifts d.Values of d(13C),d(15N),d(17O) and d(19O)shifts are given relative to gaseous CH4,NH3, H2O,and HF respectively. Due to error cancellations,the prediction of chemical shifts by MP2is signi?cantly better than that of magnetic sheildings.MAD for13C from MP2is around2.7ppm; while that for15N,17O,19F is around10.0ppm,leading to an overall MAD of around4.9ppm.RHF does not ben- e?t much from the e?ect of error cancellations.MAD for 13C from RHF is around6.5ppm;while that for15N, 17O,19F is around35.6ppm,leading to an overall MAD of15.2ppm.Deteriorated by errors in the prediction of the molecular geometry,RHF is concluded to be obsolete for routine application of NMR chemical shift calculations. Table2reveals that the prediction of chemical shifts by DFT is generally better than the prediction of the corre-sponding magnetic sheildings.In agreement with Cheese-man et al.[9],our calculations con?rmed that the performance of some commonly used functionals,such as PBEPBE(MAD(tot)=8.4)and B3LYP(MAD(tot)=6.1) is now better than that of RHF for the prediction of chem-ical shifts,although it is not as good as MP2. MAD for13C chemical shifts from PBE0is only 2.6ppm,competitive to MP2.However,PBE0and some other DFT functionals(BLYP and PBE)are signi?cantly inferior to MP2for the chemical shift prediction of other heteroatoms of15N,17O,19F.Overall,BLYP is too poor to be recommended for the calculations of NMR shifts. Most signi?cantly,we see that OPBE and OLYP are the overall winners among the DFT methods examined here. MAD for13C chemical shifts from OPBE is around 2.3ppm;while that for15N,17O,19F is around12.6ppm, leading to an overall MAD of5.4ppm,being competitive or even better in some cases than MP2. For RHF,BLYP and PBE0,the maximum errors for the prediction of chemical shifts occur at F2,being242.3, 106.6and86.6ppm,respectively.The same are true for B3LYP,PBEPBE,OPBE and MP2,although the situa-tions are less severe,such that MADs are19.3,41.1,43.4 and39.2ppm,respectively.OLYP performs most satisfac-torily.MAD for F2is about15.6ppm,although the max-imum error occurs at19F in CH3F(30.1),leaving room for further improvement. 3.4.Rovibrational e?ects Fundamentally,for the evaluation of NMR constants,it is essential to di?erentiate between the experimental r0,d0 values which refer to vibrating and rotating molecules at about300K,and the calculated r e,d e values referring to the frozen r e geometry at0K.Nevertheless,it is computa-tionally non-trivial to include the rovibrational correction [33,34].Therefore we made a direct comparison between the calculated r e,d e values and the experimental r0,d0val-ues in the previous sections as was done in some literatures [9,10,13]. For CH4,the experimental shieldings after and before the rovibrational correction are195.1(r0[28])and 198.4ppm(r e[6]),respectively.OPBE leads to194.7,with the corresponding MADs of0.4and4.3ppm.Deviation also increases by4ppm for C2H6if the rovibrational e?ects are considered[6](see Fig.1).For C2H4and C2H2,the rovibrational correction will increase the shieldings by3.5 and3.1ppm[6],respectively.As the OPBE data are more positive than the experimental r0by 1.1and 2.5ppm, respectively,the corresponding MADs are 2.4and 0.6ppm when the corrected experimental r e is used as the reference.For C6H6,the OPBE data is too shielded by6.1ppm as compared to the experimental r0,leading 386Y.Zhang et al./Chemical Physics Letters421(2006)383–388 to MAD of only2.7ppm as compared to the rovibration corrected r e value[34].From Fig.1,it is clear that the rovi-brational correction systematically increase the shielding, making r e>r0.MP2has a tendency of overshielding,lead-ing to the predicted r e,which is more close to the experi-mental r e than to the experimental r0.DFT,taking BLYP as a typical example,has a tendency of deshielding, leading to the predicted r e,which is more close to the experimental r0than to the experimental r e.OPBE is a compromise,being comparable to both the experimental r e and r0. Fig.2shows the predicted shifts by BLYP,OPBE and MP2as compared to the experimental d e and d0.As BLYP leads to too deshielded nucleus,the predicted shifts are thus too high.The errors are always higher as compared to the experimental d e than to the experimental d0.On the contrary,MP2leads to too shielded nucleus,the pre-dicted shifts are thus too small.The errors are always smal-ler as compared to the experimental d e than to the experimental d0.Again we see that OPBE is impressively good.In fact,as the deshielding e?ect is least severe in OPBE as compared to other DFT methods,changing refer-ence to the rovibration corrected r e and d e does not change the conclusion that OPBE is the best GGA functional, optimized based on energy criteria,currently available for the NMR prediction. Y.Zhang et al./Chemical Physics Letters421(2006)383–388387 4.Conclusion As a pragmatic tool of routine use,we examine the per-formance for NMR constant calculation of each method at its own optimized molecular geometry,making a direct comparison between the calculated r e,d e values and the experimental r0,d0values. Our data present here reveal that the performance of each method shows a considerable di?erence in the predic-tion of magnetic shieldings and chemical shifts.Although RHF is of similar quality in the calculations of both mag-netic shieldings and chemical shifts,MP2exhibits much higher accuracy for chemical shifts than magnetic shiel-dings.Errors of each DFT method are generally smaller in the prediction of chemical shifts.The commonly used BLYP functional is poor for the calculations of NMR con-stants.We conclude that OPBE is the overall winners in the prediction of both magnetic shieldings and chemical shifts. This is the recommended method. Comparing data between BLYP and OLYP;as well as PBEPBE and OPBE,highlights the importance of the role that the exchange functional plays;whereas contrasting the performance between OLYP and OPBE uncovers the sig-ni?cance that the correlation functional possesses.Simi-larly,comparing data between B3LYP and BLYP,as well as PBE0and PBEPBE,signi?es how hybridization scheme helps to enhance the accuracy.More systematic investigation is under way along this direction. Acknowledgements This work is supported by NSFC(20525311,20533030, 20021002,20423002),the Ministry of Science and Technol-ogy(2004CB719902,2001CB610506),TRAPOYT from the Ministry of Education. Appendix A.Supplementary data Supplementary data associated with this article can be found,in the online version,at doi:10.1016/j.cplett.2006. 01.095.References [1]T.Helgaker,M.Jaszunski,K.Ruud,Chem.Rev.99(1999)293. [2]W.Koch,M.C.Holthausen,A Chemist’s Guide to Density Func- tional Theory,Wiley,New York,2000. [3]J.Gauss,J.Chem.Phys.99(1993)3629. [4]S.M.Cybulski,D.M.Bishop,J.Chem.Phys.106(1997)4082. [5]J.Gauss,J.F.Stanton,J.Chem.Phys.103(1995)3561. [6]A.A.Auer,J.Gauss,J.F.Stanton,J.Chem.Phys.118(2003)10407. [7]C.van Wullen,W.Kutzelnigg,J.Chem.Phys.104(1996)2330. [8]P.J.Wilson,R.D.Amos,N.C.Handy,Chem.Phys.Lett.312(1999) 475. [9]J.R.Cheeseman,G.W.Trucks,T.A.Keith,M.J.Frisch,J.Chem. Phys.104(1996)5497. [10]C.Adamo,V.Barone,Chem.Phys.Lett.298(1998)113. [11]G.Magyarfalvi,P.Pulay,J.Chem.Phys.119(2003)1350. [12]S.Patchkovskii,J.Autschbach,T.Ziegler,J.Chem.Phys.115(2001) 26. [13]V.G.Malkin,O.L.Malkina,M.E.Casida,D.R.Salahub,J.Am. Chem.Soc.116(1994)5898. [14]A.M.Lee,N.C.Handy,S.M.Colwell,J.Chem.Phys.103(1995)10095. [15]A.J.Cohen,Q.Wu,W.T.Yang,Chem.Phys.Lett.399(2004)84. [16]B.Mothana,F.Q.Ban,R.J.Boyd,Chem.Phys.Lett.401(2005)7. [17]T.W.Keal,D.J.Tozer,T.Helgaker,Chem.Phys.Lett.391(2004)374. [18]A.D.Becke,Phys.Rev.B38(1988)3098. [19]J.P.Perdew,K.Burke,M.Ernzerhof,Phys.Rev.Lett.77(1996)3865. [20]C.Lee,W.Yang,R.G.Parr,Phys.Rev.B37(1988)785. [21]A.J.Cohen,N.C.Handy,Chem.Phys.Lett.316(2000)160. [22]A.D.Becke,J.Chem.Phys.98(1993)5648. [23]R.Ditch?eld,J.Chem.Phys.56(1972)5688. [24]G AUSSIAN03,Revision B.3,Gaussian Inc.,Pittsburgh,PA,2003. [25]D.J.DeFrees,B.A.Levi,S.K.Pollack,W.J.Hehre,J.S.Binkley,J.A. Pople,J.Am.Chem.Soc.101(1979)4085. [26]J.H.Callonion,E.Hirota,K.Kuchitsu,https://www.wendangku.net/doc/8a11604821.html,?erty,A.G.Maki, C.S.Pote,in:Landolt–Bornstein,Numerical Data and Function Relationships in Science and Technology,in:K.H.Hellwege(Ed.), New Series,Structure Data on Free Polyatomic Molecules,vol.7, Springer-Verlag,West Berlin,1976. [27]L.Olsson,D.Cremer,J.Phys.Chem.100(1996)16881. [28]A.K.Jameson,C.J.Jameson,Chem.Phys.Lett.134(1987)461. [29]C.J.Jameson,A.K.Jameson,D.Oppusungu,S.Wille,P.M.Burrell, J.Chem.Phys.74(1981)81. [30]R.E.Wasylishen,D.L.Bryce,J.Chem.Phys.117(2002)10061. [31]W.Makulski,K.Jackowski,Chem.Phys.Lett.341(2001)369. [32]C.J.Jameson,A.K.Jameson,P.M.Burrell,J.Chem.Phys.73(1980) 6013. [33]P.-O.A?strand,K.Ruud,Phys.Chem.Chem.Phys.5(2003)5015. [34]K.Ruud,P.-O.A?strand,P.R.Taylor,J.Am.Chem.Soc.123(2001) 4826. 388Y.Zhang et al./Chemical Physics Letters421(2006)383–388 (一)有关蛋白质和核酸计算: [注:肽链数(m);氨基酸总数(n);氨基酸平均分子量(a);氨基酸平均分子量(b);核苷酸总数(c);核苷酸平均分子量(d)]。 1.蛋白质(和多肽):氨基酸经脱水缩合形成多肽,各种元素的质量守恒,其中H、O参与脱水。每个氨基酸至少1个氨基和1个羧基,多余的氨基和羧基来自R基。 ①氨基酸各原子数计算:C原子数=R基上C原子数+2;H原子数=R基上H原子数+4;O原子数=R 基上O原子数+2;N原子数=R基上N原子数+1。 ②每条肽链游离氨基和羧基至少:各1个;m条肽链蛋白质游离氨基和羧基至少:各m个; ③肽键数=脱水数(得失水数)=氨基酸数-肽链数=n—m ; ④蛋白质由m条多肽链组成:N原子总数=肽键总数+m个氨基数(端)+R基上氨基数; =肽键总数+氨基总数≥肽键总数+m个氨基数(端); O原子总数=肽键总数+2(m个羧基数(端)+R基上羧基数); =肽键总数+2×羧基总数≥肽键总数+2m个羧基数(端); ⑤蛋白质分子量=氨基酸总分子量—脱水总分子量(—脱氢总原子量)=na—18(n—m); 2.蛋白质中氨基酸数目与双链DNA(基因)、mRNA碱基数的计算: ①DNA基因的碱基数(至少):mRNA的碱基数(至少):蛋白质中氨基酸的数目=6:3:1; ②肽键数(得失水数)+肽链数=氨基酸数=mRNA碱基数/3=(DNA)基因碱基数/6; ③DNA脱水数=核苷酸总数—DNA双链数=c—2; mRNA脱水数=核苷酸总数—mRNA单链数=c—1; ④DNA分子量=核苷酸总分子量—DNA脱水总分子量=(6n)d—18(c—2)。 mRNA分子量=核苷酸总分子量—mRNA脱水总分子量=(3n)d—18(c—1)。 ⑤真核细胞基因:外显子碱基对占整个基因中比例=编码的氨基酸数×3÷该基因总碱基数×100%;编码的氨基酸数×6≤真核细胞基因中外显子碱基数≤(编码的氨基酸数+1)×6。 3.有关双链DNA(1、2链)与mRNA(3链)的碱基计算: ①DNA单、双链配对碱基关系:A1=T2,T1=A2;A=T=A1+A2=T1+T2,C=G=C1+C2=G1+G2。A+C=G+T=A+G=C+T=1/2(A+G+C+T);(A+G)%=(C+T)%=(A+C)%=(G+T)%=50%;(双链DNA两个特征:嘌呤碱基总数=嘧啶碱基总数) DNA单、双链碱基含量计算:(A+T)%+(C+G)%=1;(C+G)%=1―(A+T)%=2C%=2G%=1―2A%=1―2T%;(A1+T1)%=1―(C1+G1)%;(A2+T2)% =1―(C2+G2)%。 ②DNA单链之间碱基数目关系:A1+T1+C1+G1=T2+A2+G2+C2=1/2(A+G+C+T); A1+T1=A2+T2=A3+U3=1/2(A+T);C1+G1=C2+G2=C3+G3=1/2(G+C); 《智能仪器原理及应用》测试题 一、填空题(每空1分共25分) 1、模拟量输入通道包括、。 2、为了将A/D转换器中的运算放大器和比较器的漂移电压降低,常采用 技术。 3、克服键抖动常采用的措施、。 4、总线收发器的作用。 5、最基本的平均滤波程序是,改进型 有、、。 6、多斜式积分器有,其优点是,还有一种是,其作用是。 7、在通用计算机上添加几种带共性的基本仪器硬件模块,通过软件来组合成各种功能的仪器或系统的仪器称为 或。 8、ADC0809,假定REF+=+5V,VREF-接地,则模拟输入为1V时,转换成的数字量为,若REF+=+2.5V,VREF-接地则模拟输入为1V时,转换成的数字量为 9、数字存储示波器可预置四种触发方 式、、、。 10、智能仪器自检方式有三种、、。 二、简答(每题5分共35分) 1、简述自由轴法测量原理。 2、系统误差的处理方法。 3、简述三线挂钩过程及作用。 4、智能仪器的设计要点。 5、若示波器屏幕的坐标刻度为8×10div,采用10位A/D,2K 存储器,则该示波器的垂直与水平分辨率各为多少? 6、简述线路反转法原理。 7、简述D/A双极性输出电路原理 三、综合 1、(20分)在一自动控制系统中,有温度、压力、流量三个待测量,试设计一测量电路,要求使用8位A/D,4位LED及相关逻辑电路。 (1)画出硬件连接图 (2)写出器件型号(CPU、A/D) (3)根据连接图,写出三通道的地址。 (4)简述测量过程。 2、(20分)下图为某一通用计数器框图 (1) 要测量10Hz的信号,试计算应选用的时标及闸门时间。 (2) 简述测量过程 (3) 其最大计数误差是多少? (4)为减小误差,应采用什么方法? 《智能仪器设计基础》试题 一、判断题(每题 2 分,共 20 分) 1. 因中值滤波满足比例不变性,所以是线性的滤波器。() 2. 基准电压Vr 的精度和稳定性影响零位误差、增益误差的校正效果。() 3. 测量获得一组离散数据建立近似校正模型,非线性校正精度与离散数据精度无关,仅与建模方法有关。() 不带横杆的矩形渡槽结构计算: 1. 槽身横向计算:沿纵向取单位长度1 m 槽身为脱离体进行计算,计算简图如图1所示。 图1.槽身横向计算简图 作用于所切取的单位长度脱离体上的荷载q 等于水重、人群荷载及槽身自重之和,除此之外,在脱离体两个侧面作用着剪力1Q 和2Q ,并由1Q 和2Q 的差值Q ?与竖向力q 保持平衡,即q Q Q Q =-=?21。 (1)人行道板计算 人行道板为一支承在侧墙上的悬臂板,计算跨长为mm a 100020012001=-=,承受的均布荷载1q 等于人群荷载加板的自重。人行道板承受的最大弯矩为: m kN a g q a q M k G k Q ?-=?+??-=+-=-= 3.11)5.21.0531.2(5.02 121212110)(γγ mm a 30=; =-=a h h 0100-30=70mm ; 0.0793*******.6103.111.226 20 =????==bh f KM c s α 468.085.00.0827211=<=--=b s ξαξ 20851300 708270.010009.6mm f h b f A y c s =???==ξ 为与侧墙钢筋协调,实配B 025@8,20201mm A =。 (2)侧墙计算 侧墙中最大计算弯矩的截面是侧墙的截面1,该处的水深为2.8m,另外为了截断部分由截面1延伸向上的竖向钢筋,距墙底1.0m 处再选取一计算截面2计算。 在工程实践中,侧墙近似的按受弯构件设计(略去轴向力影响)。侧墙底端的最大弯矩为(弯矩符号以槽壁外侧受拉为正): 截面1配筋: m kN a q H M ?-=+???-=+-=39.73.111.02.8106 12161321131)()(γ mm a 30=;=-=a h h 0300-30=270mm ;mm b 0100=; 0.056727010009.61039.71.026 20 =????==bh f KM c s α 468.085.00.0584211=<=--=b s ξαξ 20504300 2700584.010009.6mm f h b f A y c s =???==ξ 取用B 125@10,2628mm A s =。 截面2配筋: m kN a q H M ?-=+-??-=+'-=12.833.1112.8106 12161321132))(()(γ mm a 30=;=-=a h h 0300-30=270mm ;mm b 0100=; 0.018327010009.61012.831.026 20 =????==bh f KM c s α 468.085.00.0185211=<=--=b s ξαξ 20160300 2700185.010009.6mm f h b f A y c s =???==ξ 取用B 025@8,20201mm A =。 抗裂校核: 计算截面取在拖承(0.2x0.2)顶边截面3处,校核水深=H 2.8-0.2=2.6m 则: 人体细胞中的核酸有两种:DNA和RNA DNA碱基:A、T、C、G,五碳糖:脱氧核糖 RNA碱基:A、U、C、G,五碳糖:核糖 所以碱基有5种:A、T、C、G、U 五碳糖有两种:核糖、脱氧核糖 核苷酸有8种:腺嘌呤核糖核苷酸(A)、鸟嘌呤核糖核苷酸(G)、胞嘧啶核糖核苷酸(C)、尿嘧啶核糖核苷酸(U)、腺嘌呤脱氧核糖核苷酸(A)、鸟嘌呤脱氧核糖核苷酸(G)、胞嘧啶脱氧核糖核苷酸(C)、胸腺嘧啶脱氧核糖核苷酸(T) 在R基上无N元素存在的情况下,N原子的数目与氨基酸的数目相等。 .肽链中氨基酸数目、肽键数目和肽链数目之间的关系:若有n个氨基酸分子缩合成m 条肽链,则可形成(n-m)个肽键,脱去(n-m)个水分子,至少有-NH2和-COOH各m个。游离氨基或羧基数=肽链条数+R基中含有的氨基或羧基数 例1.谷胱甘肽(分子式C10H17O6N3S)是存在于动植物和微生物细胞中的一种重要的三肽,它是由谷氨酸(C5H9NO4)、甘氨酸(C2H5O2)和半胱氨酸缩合而成,则半胱氨酸可能的分子式为( ) A.C3H3NS B. C3H5NS C. C3H7O2NS D. C3H3O2NS 解析: 谷胱甘肽是由3个氨基酸通过脱去2分子水缩合而成的三肽。因此,这3个氨基酸分子式之和应等于谷胱甘肽分子式再加上2个水分子,即C10H17O6N3S+2H2O=C10H21O8N3S 。故C10H21O8N3S - C5H9NO4 -C2H5NO2 =C3H7O2NS(半胱氨酸)。 参考答案:C 点拨:掌握氨基酸分子的结构通式以及脱水缩合反应的过程是解决此类计算题的关键。 二、有关蛋白质中肽键数及脱下水分子数的计算例2. 人体内的抗体IgG是一种重要的免疫球蛋白,由4条肽链构成,共有m个氨基酸,则该蛋白质分子有肽键数( ) A.m 个B. (m+1)个 C.(m-2)个 D.(m-4)个 参考答案:D 点拨:m个氨基酸分子脱水缩合成n条多肽链时,要脱下(m-n)个水分子,同时形成(m-n) 1计算化学概述 计算化学在最近十年中可以说是发展最快的化学研究领域之一。究竟什么是计算化学呢?由于其目前在各种化学研究中广泛的应用, 我们并不容易给它一个很明确的定义。简单的来说, 计算化学是根据基本的物理化学理论通常指量子化学、统计热力学及经典力学及大量的数值运算方式研究分子、团簇的性质及化学反应的一门科学。最常见到的例子是以量子化学理论和计算、分子反应动力学理论和计算、分子力学及分子动力学理论和计算等等来解释实验中各种化学现象,帮助化学家以较具体的概念来了解、分析观察到的结果。对于未知或不易观测的化学系统, 计算化学还常扮演着预测的角色, 提供进一步研究的方向。除此之外, 计算化学也常被用来验证、测试、修正、或发展较高层次的化学理论。同时准确或有效率计算方法的开发创新也是计算化学领域中非常重要的一部分。简言之, 计算化学是一门应用计算机技术, 通过理论计算研究化学反应的机制和速率, 总结和预见化学物质结构和性能关系的规律的学科。如果说物理化学是化学和物理学相互交叉融合的产物, 那么计算化学则是化学、计算机科学、物理学、生命科学、材料科学以及药学等多学科交叉融合的产物, 而化学则是其中的核心学科。近二十年来, 计算机技术的飞速发展和理论方法的进步使理论与计算化学逐渐成为一门新兴的学科。今天、理论化学计算和实验研究的紧密结合大大改变了化学作为纯实验科学的传统印象, 有力地推动了化学各个分支学科的发展。而且, 理论与计算化学的发展也对相关的学科如纳米科学和分子生物学的发展起到了巨大的推动作用。 2计算化学的产生、发展、现状和未来 2.1计算化学的产生 计算化学是随着量子化学理论的产生而发展起来的, 有着悠久历史的一门新兴学科。自上个世纪年代量子力学理论建立以来, 许多科学家曾尝试以各种数值计算方法来深人了解原子与分子之各种化学性质。然而在数值计算机广泛使用之前, 此类的计算由于其复杂性而只能应用在简单的系统与高度简化的理论模型之中, 所以, 即使是在此后的数十年里, 计算化学仍是一门需具有高度量子力学与数值分析素养的人从事的研究, 而且由于其庞大的计算量, 绝大部分的 目录 1. 工程概况.............................................. 错误!未定义书签。2.槽身纵向内力计算及配筋计算............................ 错误!未定义书签。 (1)荷载计算..........................................错误!未定义书签。 (2)内力计算..........................................错误!未定义书签。 (3)正截面的配筋计算..................................错误!未定义书签。 (4)斜截面强度计算....................................错误!未定义书签。 (5)槽身纵向抗裂验算..................................错误!未定义书签。3.槽身横向内力计算及配筋计算............................ 错误!未定义书签。 (1)底板的结构计算....................................错误!未定义书签。 (2)渡槽上顶边及悬挑部分的结构计算 ....................错误!未定义书签。 (3)侧墙的结构计算....................................错误!未定义书签。 (4)基地正应力验算....................................错误!未定义书签。 1. 工程概况 重建渡槽带桥,原渡槽后溢洪道断面下挖,以满足校核标准泄洪要求。目前,东方红干渠已整修改造完毕,东方红干渠设计成果显示,该渡槽上游侧渠底设计高程为165.50m,下游侧渠底设计高程为165.40m。本次设计将现状渡槽拆除,按照上述干渠设计底高程,结合溢洪道现状布置及底宽,在原渡槽位重建渡槽带桥,上部桥梁按照四级道路标准,荷载标准为公路-Ⅱ级折减,建筑材料均采用钢筋砼,桥面总宽5m。 现状渡槽拆除后,为满足东方红干渠的过流要求及溢洪道交通要求,需重建跨溢洪道渡槽带桥。新建渡槽带桥轴线布置于溢洪道桩号0+,同现状渡槽桩号,下底面高程为165.20m,满足校核水位+0.5m超高要求,桥面高程167.40m,设计为现浇结合预制混凝土结构,根据溢洪道设计断面,确定渡槽带桥总长51m,8.5m×6跨。上部结构设计如下:渡槽过水断面尺寸为×1.6m,同干渠尺寸,采用C25钢筋砼,底及侧壁厚20cm,顶壁厚30cm,筒型结构,顶部两侧壁水平挑出1.25m,并在顺行车方向每隔2m设置一加劲肋,维持悬挑板侧向稳定,桥面总宽5m,路面净宽4.4m,设计荷载标准为公路-Ⅱ级折减,两侧设预制C20钢筋砼栏杆,基础宽0.5m。下部结构设计如下:下部采用C30钢筋混凝土双柱排架结构,并设置横梁, 由于地基为砂岩,基础采用人工挖孔端承桩,尺寸为×1.2m,基础深入岩层弱风化层1.0m,盖梁尺寸为4××1.2m。 2.槽身纵向内力计算及配筋计算 根据支承形式,跨宽比及跨高比的大小以及槽身横断面形式等的不同,槽身应力状态与计算方法也不同,对于梁式渡槽的槽身,跨宽比、跨高比一般都比较大,故可以按 “蛋白质计算”专题讲练 在高中生物学中,涉及蛋白质各种因素之间的数量关系比较复杂,是学生学习中的重点和难点,也是高考的考点与热点。因此,在复习时牢牢掌握氨基酸分子的结构通式以及脱水缩合反应的过程,恰当的运用相关公式是解决问题的关键。现将与蛋白质相关的计算公式及典型例题归析如下,以便复习参考。 一、有关蛋白质计算的公式汇总 ★★规律1:有关氨基数和羧基数的计算 ⑴蛋白质中氨基数=肽链数+R基上的氨基数=各氨基酸中氨基的总数-肽键数; ⑵蛋白质中羧基数=肽链数+R基上的羧基数=各氨基酸中羧基的总数-肽键数; ⑶在不考虑R基上的氨基数时,氨基酸脱水缩合形成的一条多肽链中,至少含有的氨基数为1,蛋白质分子由多条肽链构成,则至少含有的氨基数等于肽链数; ⑷在不考虑R基上的羧基数时,氨基酸脱水缩合形成的一条多肽链中,至少含有的羧基数为1,蛋白质分子由多条肽链构成,则至少含有的羧基数等于肽链数。 ★★规律2:蛋白质中肽键数及相对分子质量的计算 ⑴蛋白质中的肽键数=脱去的水分子数=水解消耗水分子数=氨基酸分子个数-肽链数; ⑵蛋白质的相对分子质量=氨基酸总质量(氨基酸分子个数×氨基酸平均相对分子质量)-失水量(18×脱去的水分子数)。 注意:有时还要考虑其他化学变化过程,如:二硫键(—S—S—)的形成等,在肽链上出现二硫键时,与二硫键结合的部位要脱去两个H,谨防疏漏。 ★★规律3:有关蛋白质中各原子数的计算 ⑴C原子数=(肽链数+肽键数)×2+R基上的C原子数; ⑵H原子数=(氨基酸分子个数+肽链数)×2+R基上的H原子数=各氨基酸中H原子的总数-脱去的水分子数×2; ⑶O原子数=肽链数×2+肽键数+R基上的O原子数=各氨基酸中O原子的总数-脱去的水分子数; ⑷N原子数=肽链数+肽键数+R基上的N原子数=各氨基酸中N原子的总数。 《计算化学》课程学习指南 计算化学学习基本要求: 在学习了化学系列基础课程之后,通过本课程的学习,掌握化学中常用的数值计算方法,并能利用计算方法来解决化学中和部分工程实践中的实际问题,学习中坚持理论与实践相结合,才能更深刻的理解与运用理论,并在解决实际问题中,掌握理论和方法,培养学习能力、实践能力和创新能力。 计算化学学习的难点: 学生学习计算化学时由于受原有化学、数学、计算机基础的制约,感到课程涉及知识面广,入门较慢。尤其是对各种化学、化工知识的综合应用及编程需要有一个熟悉的过程。坚持一定会有收获! 计算化学的研究方法: 传统意义上的计算化学要完成的任务一般包括以下几个方面: 1.量子结构计算,分子从头计算(Schrodinger方程的精确解)、半经验计算(Schrodinger方程的估计解)和分子力学计算(根据分子参数计算),属于量子化学和结构化学范畴; 2.物理化学参数的计算,包括反应焓、偶极矩、振动频率、反应自由能、反应速率等的理论计算,一般属于统计热力学范畴; 3.化学过程模拟和化工过程计算等。 但是随着科学的发展,要界定计算化学的范围是很困难的,因为它是化学学科现代化过程中新的生长点,它与迅速崛起的高科技关系密切,深受当今计算机及其网络技术飞速发展的影响,正处在迅速发展和不断演变之中,研究的侧重点也因研究者及其所处的学术环境、原有基础和人员的知识背景而异。在今后的一段时期内,计算机辅助结构解析、分子设计和合成路线设计将是计算化学的主题。尽管实际上计算化学覆盖的面还要广得多,比较公认的研究领域至少有:1.化学数据挖掘(Data mining); 2.化学结构与化学反应的计算机处理技术; 3.计算机辅助分子设计; 4.计算机辅助合成路线设计; 5.计算机辅助化学过程综合与开发; 6.化学中的人工智能方法等。 无论计算化学涉及的内容多么广泛,其核心依然是数值计算问题。 本课程主要学习利用计算机解化学中的数值计算问题,一般包括以下几个步骤: 1.对所要解决的问题进行分析,将化学问题转变为数学模型,选择所需的计算方法; 问题分析是完成计算任务的基础,包括对问题所含物理化学意义的清楚认识。在进行数值计算时要量纲明确,保证计算步骤分解准确。采用的数学理论正确、计算方法合理有效。 2.写出解决问题的程序框图 根据分析结果给出程序框图是编写程序的基础和关键。写出清晰、流畅、准确的程序框图是任何计算机语言编写程序的必要步骤。程序框图的绘制要根据计算机运算的特点和编写代码程序的需要。 3.代码程序的编写 选择一种合适的计算机语言,运用该种语言将上述程序框图写成计算机程序(高级程序)。由于一种计算机语言往往有不同版本,适合于不同的编译平台,彩的程序代码要符合该编译平台的规范。 4.程序的调试和编译 一个计算机程序编写完成后,一般需要通过编译、调试和修改步骤,构成计算机可以识别的代码集,并找出问题,加以完善。编译和高度的方法依据不同的程序编译平台会略有不同。 5.试算分析,输出结果 调试得到执行程序后,用已知的算例去试算检查,分析结果正确无误码,才能用于未知的算例。 蛋白质、核酸的计算 题型精讲 题型1、蛋白质的相关计算 1.某直链多肽的分子式为C22H34O13N6,其彻底水解后共产生了以下3种氨基酸,相关叙述正确的是 A.该直链多肽含一个游离氨基和一个游离羧基 B.该多肽没有经过折叠加工,所以不能与双缩脲试剂反应 C.合成1分子该物质将产生6个水分子 D.每个该多肽分子水解后可以产生3个谷氨酸 【答案】D 2.现有氨基酸800个,其中氨基总数为810个,羧基总数为808个,则由这些氨基酸合成的含有2条肽链的蛋白质共有肽键、氨基和羧基的数目依次为 A.798、2和2 B.799、11和9 C.799、1和1 D.798、12和10 【答案】D 题型2、核酸的相关计算 1.某双链DNA分子中,鸟嘌呤与胞嘧啶之和占全部碱基的比例为a,其中一条链上鸟嘌呤占该链全部碱基的比例为b,则互补链中鸟嘌呤占整个DNA分子碱基的比例为 A.(a/2)—(b/2) B.a—b C.(a—b)/(1—a) D.b—(a/2) 【答案】A 2.已知1个DNA分子中有4000个碱基对,其中胞嘧啶有2200个,这个DNA分子中应含有的脱氧核苷酸的数目和腺嘌呤的数目分别是 A.4 000和900个 B.4 000和1 800个 C.8 000和1800个 D.8 000和3 600个 【答案】C 同步练习 1.一条肽链的分子式为C22H34O13N6,其水解产物中只含有下列3种氨基酸。下列叙述错误的是 A.合成1个C22H34O13N6分子将产生5个水分子 B.在细胞中合成1个C22H34O13N6分子要形成5个肽键 C.1个C22H34O13N6分子完全水解后可以产生3个谷氨酸 D.1个C22H34O13N6分子中存在1个游离的氨基和3个游离的羧基 【答案】D 2.N个氨基酸组成了M个肽,其中有Z个是环状肽,据此分析下列表述错误的是A.M个肽一定含有的元素是C、H、O、N,还可能含有S B.M个至少含有的游离氨基数和游离羧基数均为M-Z C.将这M个肽完全水解为氨基酸,至少需要N-M+Z个水分子 D.这M个肽至少含有N-M+Z个O原子 【答案】D 3.有一种“十五肽”的化学式为C x H y N z O d S e(z>15,d>16)。已知其彻底水解后得到下列几种氨基酸:下列有关说法中不正确的是 A.水解可得e个半胱氨酸 B.水解可得(d-16)/2个天门冬氨酸 C.水解可得z-15个赖氨酸 D.水解时消耗15个水分子 【答案】D 4.某一DNA分子含有800个碱基对,其中含A为600个。该DNA分子含G的脱氧核苷酸的个数是 A.600 B.800 一、基本资料 1.1工程等别 根据《水利水电工程等级划分及洪水标准》(SL252-2000)、《灌溉与排水工程设计规范》(GB50288-99)和《村镇供水工程技术规范》(SL687—2014)的规定,工程设计引水流量为3.9m3/s,供水对象为一般,确定本项目为Ⅳ等小(1)型工程。主要建筑物等级为4等,次要建筑物等级为5等,临时建筑物等级为5等。 渡槽过水流量≤5m3/s,故渡槽等级均为5级。 1.2设计流量及上下游渠道水力要素 正常设计流量1.83m3/s,加大流量2.29 m3/s。 1.3渡槽长度 槽身长725m,进出口总水头损失0.5m。 1.4地震烈度 工程区位于安陆市北部的洑水镇、接官乡和赵鹏镇三个乡镇,属构造剥蚀丘岗地貌。根据国家标准1:400万《中国地震动参数区划图》(GB18306-2001),工程区地震动峰值加速度为0.05g,地震动反应谱特征周期为0.35s,相应的地震基本烈度小于Ⅵ度,建筑物不设防。 1.5水文气象资料 安陆市属亚热带季风气候区,春秋短,冬夏长,四季分明,兼有南北气候特点。年最高气温40.5℃,最低气温-15.3℃,多年平均气温15.9℃。年日照时数1920—2440h,日照率49%,居邻近各县(市)之冠。太阳总辐射年平均112千卡/cm2,年际变化不大,4-10月辐射量占全年的71.43%。10℃以上积温为4486—4908℃。多年平均无霜期246d。 境内多年平均降雨量1117mm,年降雨量很不稳定,最多年份可达1772.6mm (1954年),最少年份只有652.9 mm(1978年),降水量年内分配很不均匀,4-10月份平均降雨量占全年降雨量的85%以上,多年平均蒸发量1587.3mm,由于降水量年际和年内间变化大,导致洪涝旱灾发生频繁。 渡槽箱形梁结构计算书(11.18) 一、槽身纵向内力计算及配筋计算 根据支承形式,跨宽比及跨高比的大小以及槽身横断面形式等的不同,槽身应力状态与计算方法也不同,对于梁式渡槽的槽身,跨宽比、跨高比一般都比较大,故可以按梁理论计算。槽身纵向按正常过水高程计算(本渡槽设计水位高程取60cm)。 图1—1 槽身横断面型式(单位:mm) 1、荷载计算 根据设计拟定,渡槽的设计标准为5级,使用年限50年所以渡槽的安全级别Ⅲ级, 则安全系数为γ =0.9(DL-T 5057 -2009规范),C30混凝土重度为γ=25kN/m3(根据水工混凝土结构设计规范DL-T 5057-2009:6.1.7条),正常运行期为持久状况,其设计状况 系数为ψ=1.0,荷载分项系数为:永久荷载分项系数γ G =1.05,可变荷载分项系数γ Q =1.20 (《水工建筑物荷载设计规范》(DL 5057 -1997规范)),结构系数为γ d =1.2(DL-T 5057 -2009规范)。 纵向计算中的荷载一般按匀布荷载考虑,包括槽身重力(栏杆等小量集中荷载也换算为匀布的)、槽中水体的重力及人群荷载。其中槽身自重、水重为永久荷载,而人群荷载 为可变荷载。 (1)槽身自重: 标准值:G 1k =γ ψγ(V 1 +2V 2 +V 3 )=0.9×1×25×(0.15× 2.3+0.7×0.25×2+1.4×0.2)=21.94(kN/m) 设计值:G 1=γ G ×g 1k =1.05×21.94=23.04(kN/m) (a )面板自重 设计值:g 1=γG γ0ψγV 1=1.05×0.9×1×25×(0.15×2.3)=8.15(kN/m ) (b )腹板自重 设计值:g 2=γG γ0ψγ2V 2=1.05×0.9×1×25×(0.25×0.7)×2=8.27(kN/m ) (c )底板自重 设计值:g 3=γG γ0ψγV 3=1.05×0.9×1×25×(1.4×0.2)=6.62(kN/m ) (2)水重:标准值:G 2k =γ0ψγV 4=0.9×9.81×1×(0.6×0.9)=4.77(kN/m ) 设计值:G 2=γG ×g 2k =1.05×4.77=5.01(kN/m ) (3)栏杆荷载: 本设计采用大理石栏杆,大理石的容重γ1=28kN/m3,缘石采用C30 混凝土预制,C25混凝土重度为γ=25kN/m 3 。 标准值:G 3k =γ0ψγ1V 5+γ0ψγV 6=0.9×1×28×2×{(0.5×0.16×0.16×5÷ 10)+0.8×0.16}+0.9×1×25×2×(0.16×0.3)=8.92(kN/m ) 设计值: G 3=γG ×g 2k =1.05×8.92=9.37(kN/m ) 根据《城市桥梁设计荷载标准》(CJJ77-98) 规范要求:桥上人行道 栏杆时,作用在栏杆扶手上的活载,竖向荷载采用1.2kN/m ;水平向外 荷载采用1.0kN/m 。两者分别考虑,不得同时作用。 标准值: Q 栏杆竖向=1.2(kN/m ) 设计值: Q 1=1.2×1.2=1.44(kN/m ) (4)人群荷载: 根据《城市桥梁设计荷载标准》(CJJ77-98) 规范要求:梁、桁、拱及其他大跨结构的人群荷载w ,可按下列公式计算,且ω值在任何情况下不得小于2.4kPa 。 当跨径或加载长度l <20m 时: 生物必修一蛋白质计算公式总结 导读:我根据大家的需要整理了一份关于《生物必修一蛋白质计算公式总结》的内容,具体内容:纵观近几年高考试题,与生物必修一蛋白质计算有关的内容进行了不同程度的考查,下面是我给大家带来的,希望对你有帮助。生物必修一蛋白质计算公式[注:肽链数(m);氨基酸总数(... 纵观近几年高考试题,与生物必修一蛋白质计算有关的内容进行了不同程度的考查,下面是我给大家带来的,希望对你有帮助。 生物必修一蛋白质计算公式 [注:肽链数(m);氨基酸总数(n);氨基酸平均分子量(a);氨基酸平均分子量(b);核苷酸总数(c);核苷酸平均分子量(d)]。 1.蛋白质(和多肽):氨基酸经脱水缩合形成多肽,各种元素的质量守恒,其中H、O参与脱水。每个氨基酸至少1个氨基和1个羧基,多余的氨基和羧基来自R基。①氨基酸各原子数计算:C原子数=R基上C原子数+2;H 原子数=R基上H原子数+4;O原子数=R基上O原子数+2;N原子数=R基上N 原子数+1。②每条肽链游离氨基和羧基至少:各1个;m条肽链蛋白质游离氨基和羧基至少:各m个; ③肽键数=脱水数(得失水数)=氨基酸数-肽链数=n—m ;④蛋白质由m条多肽链组成:N原子总数=肽键总数+m个氨基数(端)+R基上氨基数; =肽键总数+氨基总数肽键总数+m个氨基数(端); O原子总数=肽键总数+2(m个羧基数(端)+R基上羧基数); =肽键总数+2×羧基总数肽键总数+2m个羧基数(端); ⑤蛋白质分子量=氨基酸总分子量—脱水总分子量(—脱氢总原子 量)=na—18(n—m); 2.蛋白质中氨基酸数目与双链DNA(基因)、mRNA碱基数的计算: ①DNA基因的碱基数(至少):mRNA的碱基数(至少):蛋白质中氨基酸的数目=6:3:1; ②肽键数(得失水数)+肽链数=氨基酸数=mRNA碱基数/3=(DNA)基因碱基数/6; ③DNA脱水数=核苷酸总数—DNA双链数=c—2; mRNA脱水数=核苷酸总数—mRNA单链数=c—1; ④DNA分子量=核苷酸总分子量—DNA脱水总分子量=(6n)d—18(c—2)。 mRNA分子量=核苷酸总分子量—mRNA脱水总分子量=(3n)d—18(c—1)。 ⑤真核细胞基因:外显子碱基对占整个基因中比例=编码的氨基酸数 ×3÷该基因总碱基数×100%;编码的氨基酸数×6真核细胞基因中外显子碱基数(编码的氨基酸数+1)×6。 3.有关双链DNA(1、2链)与mRNA(3链)的碱基计算: ①DNA单、双链配对碱基关系:A1=T2,T1=A2;A=T=A1+A2=T1+T2, C=G=C1+C2=G1+G2。 A+C=G+T=A+G=C+T=1/2(A+G+C+T);(A+G)%=(C+T)%=(A+C)%=(G+T)%=50%;(双链DNA两个特征:嘌呤碱基总数=嘧啶碱基总数) DNA单、双链碱基含量计算: (A+T)%+(C+G)%=1;(C+G)%=1―(A+T)%=2C%=2G%=1―2A%=1―2T%;(A1+T1)% =1―(C1+G1)%;(A2+T2)% 紫外可见分光光度计的原理与应用 1.原理 物质的吸收光谱本质上就是物质中的分子和原子吸收了入射光中的某些特定波长的光能量,相应地发生了分子振动能级跃迁和电子能级跃迁的结果。由于各种物质具有各自不同的分子、原子和不同的分子空间结构,其吸收光能量的情况也就不会相同,因此,每种物质就有其特有的、固定的吸收光谱曲线,可根据吸收光谱上的某些特征波长处的吸光度的高低判别或测定该物质的含量,这就是分光光度定性和定量分析的基础。分光光度分析就是根据物质的吸收光谱研究物质的成分、结构和物质间相互作用的有效手段。 紫外可见分光光度法的定量分析基础是朗伯-比尔 (Lambert-Beer)定律。即物质在一定浓度的吸光度与它的吸收介质的厚度呈正比 2 应用 2.1 检定物质 根据吸收光谱图上的一些特征吸收,特别是最大吸收波长虽ax 和摩尔吸收系数是检定物质的常用物理参数。这在药物分析上就有着很广泛的应用。在国内外的药典中,已将众多的药物紫外吸收光谱的最大吸收波长和吸收系数载入其中,为药物分析提供了很好的手段。 2.2 与标准物及标准图谱对照 将分析样品和标准样品以相同浓度配制在同一溶剂中,在同一条 件下分别测定紫外可见吸收光谱。若两者是同一物质,则两者的光谱图应完全一致。如果没有标样,也可以和现成的标准谱图对照进行比较。这种方法要求仪器准确,精密度高,且测定条件要相同。 2.3 比较最大吸收波长吸收系数的一致性 2.4 纯度检验 2.5 推测化合物的分子结构 2.6 氢键强度的测定 实验证明,不同的极性溶剂产生氢键的强度也不同,这可以利用紫外光谱来判断化合物在不同溶剂中氢键强度,以确定选择哪一种溶剂。 2.7 络合物组成及稳定常数的测定 2.8 反应动力学研究 2.9 在有机分析中的应用 有机分析是一门研究有机化合物的分离、鉴别及组成结构测定的科学,它是在有机化学和分析化学的基础上发展起来的综合性学科。 原子吸收分光光度计工作原理 一、设计基本资料 1.1工程综合说明 根据丰田灌区渠系规划,在灌区输水干渠上需建造一座跨越小禹河的渡槽,由左岸向右岸输水。渡槽槽址及渡槽轴线已由规划选定(见渡槽槽址地形图)。渡槽按4级建筑物设计。 1.2气候条件 槽址地区位于大禹乡境内,植被良好。夏季最高气温36℃,冬季最低气温-32℃,最大冻层深度1.7m。地区最大风力为9级,相应风速v = 24 m / s。 1.3水文条件 根据水文实测及调查,槽址处小禹河平时基流量在0.2—0.4 m3/S之间,有时断流。洪水多发生在每年7、8月份;春汛一般发生在每年3月上旬,但流量不大。经水文计算,槽址处设计洪水位为1242.41m,相应流量 Q = 698 m3/S;最高洪水位为1243.83m,相应流量 Q = 1075 m3/S。据调查,洪水中漂浮物多为树木、牲畜,最大不超过400 kg。在春汛中无流冰发生。 槽址处小禹河两岸表层为壤土分布;表层以下及河床为砂卵石分布(见渡槽轴线断面图)。地基基本承载力壤土为34 t / m2;砂卵石为43 t / m2。 1.4工程所需材料要求 在建材方面,距槽址50km大禹镇有县办水泥厂一座,水泥质量合格,可满足渡槽建造水泥需要;槽址附近有大量砂石骨料分布,质量符合混凝土拌制需要,运距均在5km以内;槽址东北禹王山有石料可供开采,运距350km。 1.5上、下游渠道资料 根据灌区渠系规划,渡槽上下游渠道坡降均为1/5000。渠道底宽按设计流量计算2.7 m,边坡1:1.5,采用混凝土板衬砌。渠道设计流量6立方米每秒, 加大流量7.5立方米每秒。渠道堤顶超高0.5m。 根据灌区渠系规划,上游渠口(左岸)水面高程加大流量时为1251.04m。下游渠口(右岸)水面高程加大流量时为1250.54m。渠口位置见渡槽槽址地形图。 2012年秋季学期《计算化学》综述 分子模拟在化学领域的应用进展 班号:10907401 学号:1090740112 姓名:贺绍飞 2012年哈尔滨工业大学 分子模拟在化学领域的应用进展 摘要:分子模拟作为一种全新的研究手段已经在化学、化工、材料、生物等领域受到了广泛的关注。本文首先对分子模拟进行了简单的介绍,然后举例详细阐述了分子模拟在石油化工领域、超临界流体领域、分子筛吸附、高分子领域以及气体膜分离领域的应用发展,最后展望了分子模拟技术的发展方向。 关键词:分子模拟、问题及发展趋势、应用发展 1.引言 分子模拟技术是随着计算机在科研中的应用而发展起来的一门新的科学,是计算机科学和基础科学相结合的产物。 20世纪80年代以来,随着计算机性能的提高以及各种计算化学方法的改进,分子模拟技术日渐成熟,并逐步发展成为人们进行科学研究的一项新的有效的工具,在化学、制药、材料等相关的工业上发挥着越来越重要的作用。 分子模拟之所以受到这样的重视,与它自身的特点和相关学科的发展是密不可分的。以前,采取的都是实验室人工合成一种新型化合物,但是有一些化合物的合成繁琐而复杂,例如具有多种旋光性的药物,每一种新的药物合成都是一个工作量巨大的实验过程,以往只能采用实验手段研究时,新药的实验过程经常持续数十年,其间经历了许多失败的实验,耗费大量的人力物力。但是,在采用分子模拟的方法后,可以通过计算机模拟的手段对实验进行大量的预先筛选,大大加快了这一研究的进程。又如在对超临界流体的研究中,分子模拟和传统的实验相比有着巨大的经济优势。 2.分子模拟简介 2.1 分子模拟的定义 分子模拟是一个广泛的概念,其包括基于量子力学的模拟和基于统计力学的模拟。前者为计算量子化学(computational quantum chemistry,简称CQC),后者主要分为两个方法,分别是分子动力学模拟(molecular dynamics,MD)和蒙特卡洛模拟(Monte Carlo,MC)[1]。三者中以计算量子化学的结果最为可靠,但是其计算量也是最大的,通常处理的体系也是比较小的.MC和MD都是基于位能函数的模拟,不同之处在于MD模拟过程与时间相关,除了和MC一样可以处理平衡性质以外,在处理传递性质等与时间相关的问题时有天然的优势,当然MD 和MC相比程序的复杂程度要高,计算的难度要大一些。 2.2 分子模拟的方法[2-7] 分子模拟的方法主要有四种:分子力学方法,分子动力学方法、蒙特卡洛方法、量子力学方法。 2.2.1 分子力学方法 分子力学法又称Force Field方法,是在分子水平上解决问题的非量子力学技术。其原理是,分子内部应力在一定程度上反映被计算分子结构的相对位能大小。分子力学法是依据经典力学的计算方法,即依据Born-Oppenheimer原理,计算中将电子的运动忽略,而将系统的能量视为原子核种类和位置的函数,这些势能函数被称为力场。分子的力场含有许多参数,这些参数可由量子力学计算或实验方法得到。该法可用来确定分子结构的相对稳定性,广泛地用于计算各类化合物的分子构象、热力学参数和谱学参数。 2.2.2 分子动力学方法 分子动力学模拟是一种用来计算一个经典多体系的平衡和传递性质的方法。 化学专业学生必备:各种仪器分析的基本原理及谱图表示方法!! 紫外吸收光谱 UV 分析原理:吸收紫外光能量,引起分子中电子能级的跃迁 谱图的表示方法:相对吸收光能量随吸收光波长的变化 提供的信息:吸收峰的位置、强度和形状,提供分子中不同电子结构的信息 荧光光谱法 FS 分析原理:被电磁辐射激发后,从最低单线激发态回到单线基态,发射荧光谱图的表示方法:发射的荧光能量随光波长的变化 提供的信息:荧光效率和寿命,提供分子中不同电子结构的信息 红外吸收光谱法 IR 分析原理:吸收红外光能量,引起具有偶极矩变化的分子的振动、转动能级跃迁 谱图的表示方法:相对透射光能量随透射光频率变化 提供的信息:峰的位置、强度和形状,提供功能团或化学键的特征振动频率拉曼光谱法 Ram 分析原理:吸收光能后,引起具有极化率变化的分子振动,产生拉曼散射谱图的表示方法:散射光能量随拉曼位移的变化 提供的信息:峰的位置、强度和形状,提供功能团或化学键的特征振动频率核磁共振波谱法 NMR 分析原理:在外磁场中,具有核磁矩的原子核,吸收射频能量,产生核自旋能级的跃迁 谱图的表示方法:吸收光能量随化学位移的变化 提供的信息:峰的化学位移、强度、裂分数和偶合常数,提供核的数目、所处化学环境和几何构型的信息 电子顺磁共振波谱法 ESR 分析原理:在外磁场中,分子中未成对电子吸收射频能量,产生电子自旋能级跃迁 谱图的表示方法:吸收光能量或微分能量随磁场强度变化 提供的信息:谱线位置、强度、裂分数目和超精细分裂常数,提供未成对电子密度、分子键特性及几何构型信息 质谱分析法 MS 分析原理:分子在真空中被电子轰击,形成离子,通过电磁场按不同m/e分离 谱图的表示方法:以棒图形式表示离子的相对峰度随m/e的变化 提供的信息:分子离子及碎片离子的质量数及其相对峰度,提供分子量,元素组成及结构的信息 气相色谱法 GC 分析原理:样品中各组分在流动相和固定相之间,由于分配系数不同而分离 工程名称: 哈密市五堡镇五堡大桥渡槽工程 设计阶段:施工阶段 渡槽计算书 计算: 日期:2015.09.01 哈密托实水利水电勘测设计有限责任公司 2015.09.01 1 基本资料 五堡大桥渡槽定为4级建筑物,设计流量Q =1.2m3/s ,加大流量Q m=1.56m3/s。, 设 渡槽总长25.6m,进口与上游改建梯形现浇砼渠道连接,出口与下游改建矩形现浇砼渠道连接。 2 渡槽选型与布置 2.1 结构型式选择 梁式渡槽的槽身是直接搁置于槽墩或槽架之上的。为适应温度变化及地基不均匀沉陷等原因而引起的变形,必须设置变形缝将槽身分为独立工作的若干节,并将槽身与进出口建筑物分开。变形缝之间的每一节槽身沿纵向是两个支点所以既起输水作用又起纵向梁作用。根据支点位置的不同,梁式渡槽有简支梁式双悬臂梁式和单悬臂梁式三种型式。 单悬臂梁式一般只在双悬臂梁式向简支梁式过渡或与进出口建筑物连接时使用。 简支梁式槽身施工吊装方便,接缝止水构造简单,但跨中弯矩较大,底板受拉对抗裂防渗不利。简支梁式槽身常用的跨度为8-15m。本设计采用简支梁式槽身,跨度取为12.8m。梁式渡槽的槽身采用钢筋混凝土结构。 2.2 总体布置 渡槽的位置选择是选定渡槽的中心线及槽身起止点的位置。本设计的渡槽的中心线已选定。具体选择时可以从以下几方面考虑: (1)槽址应尽量选在地质良好、地形有利和便于施工的地方,以便缩短槽身长度、减少工程量、降低墩架高度; (2)槽轴线最好成一直线,进口和出口避免急转弯,否则将恶化水流条件,影响正常输水; (3)跨越河流的渡槽,槽轴线应与河道水流方向尽量成正交,槽址应位于河床及岸坡稳定、水流顺直的地段,避免位于河流转弯处; 2.3 结构布置 根据渠系规划确定,选用钢筋混凝土简支梁式渡槽进行输水,槽身采用带拉杆的矩形槽,支承结构采用单排架型式,两立柱之间设横梁,基础采用整体板式基础支撑排架。渡槽全长25.6m,采用等跨布置方案,一跨长度为12.8m。进出口均用混凝土建造。 从计算化学到生物学 杨金才 1501110432 尽管我是生物背景,但我所用的分子模拟方法却多是由计算化学家所建立的,然 后被应用于生物学领域。在计算化学领域主要荣获两次诺贝尓化学奖,第一次是1998年,用于表彰WalterKohn发展了密度泛函理论和John Pople发展了量子化学(QM)计算方法;第二次是2013年,授予Martin Karplus, Michael Levitt 和AriehWarshel,获奖理由 是“为复杂化学系统创立了多尺度模型”。如果说1998年获奖的量子化学计算方法使计算小分子化学体系成为可能,那2013年获奖的分子动力学计算方法则为计算生物大分子的行为提供了有力的工具,并且真正应用于揭示生物大分子功能和药物设计等实际应用 中来,理论化学终于走向了应用。 毫无疑问,量子力学计算方法的发展是极其重要的,但由于其计算量巨大,难以 应用于生物学大分子。因为如果采用量子力学计算方法算蛋白的运动轨迹,或许算100 年也不一定能算出来,对于生物大分子的计算,我们需要的是能在可以接受的时间内获 得有意义的结果。这就要求对体系作一定的近似以减少计算量,同时又最大可能地揭示 其生物学特性。而Martin Karplus在这方面做出了重要的工作,并开辟了用分子模拟解 决生物问题这一全新领域。 时间回到1950年,20岁的Martin Karplus,刚从哈佛大学毕业,当时他有两个选择,学化学或者学生物。经过美国理论物理学家、美国“原子弹之父” Robert Oppenheimer的推荐,他最终选择了生物学。于是Karplus到了西海岸的加州大学攻读生 物博士学位,师从Linus Carl Pauling。Pauling是著名美国化学奖,是量子化学和结构生 物学的先驱之一。他是唯一的一位两次独自获得诺贝尔奖的人。一次是1954年的诺贝尔化学奖,表彰其将量子力学应用于化学键的研究,深刻改变了我们对化学键的认识。于1935年出版了《量子力学导论——及其在化学中的应用》,这是历史上第一本以化学家 为读者的量子力学教科书。另一次则因参与反战反核获得1964年诺贝尔和平奖。Pauling还根据晶体衍射图,于1951年最早提出了蛋白质α螺旋结构模型。有科学史学 者认为沃森和克里克提出的DNA双螺旋结构模型就是受到了鲍林的影响。Pauling在量 子化学和结构生物学上的成就深刻影响了Karplus,“我的导师鲍林对我的科学研究产生了非常大的影响。”他说。正是在这样的学术背景下,Karplus开创了自己的领域。高中生物蛋白质部分计算题
智能仪器原理及应用
渡槽课程设计--三峡大学版
高一生物蛋白质计算总结
1计算化学概述
渡槽结构计算书
高中生物蛋白质相关计算专题
计算化学学习指南
高三高考生物重难点专题复习:蛋白质、核酸的计算
U型渡槽结构计算书
渡槽箱形梁结构计算书(11.18)
生物必修一蛋白质计算公式总结
各种仪器原理及应用
渡槽设计计算书
计算化学论文综述上交版
各种化学仪器的原理使用
矩形渡槽设计计算说明书
从计算化学到生物学_计算生物学的起源